Creating the ESPOIR Control file (filename.dat)
Before running ESPOIR, the Espoir control file has to be created. It is possible to reuse a file but you may miss-interpret the meaning of a parameter causing unexpected results. Initially it would be better to generate the file from scratch using the PRESPOIR program.ESPOIR and PRESPOIR use a DOS like interface so you would normally run it from the command line (go into the subdirectory where the files are located and call the programs the old fashioned way)
ESPOIR is developing all the time so it is good to check for updates. Tonight's update (17th April 2000) is that ESPOIR has Fullprof like comments in the control file (filename.dat) - hurrah! You may have to fiddle around to get ESPOIR working to your satisfaction in solving structures successfully.
To make a start, open up a DOS Command prompt and enter the directory with the data. Type prespoir to be given the following window.
- Remember: use the Fobs HKL data NOT the Fsquared HKL data!
(have already made this mistake myself - Lachlan) - (Note: using the Windows cut and paste can save a lot of problems, expecially minimising
the introduction of typographical errors)
- (Note: If using the "regenerated pseudo-pattern", you will be prompted to give the U,V,W from
the Le Bail fit. Please have this information handy. The following HKL file was Le Bail fitted using
Fullprof)
- When prompted, give the filename (in this case cime)
- When prompted, give an identifier (in this case cime)
- Give the cell constants (you can use a Windows cut and paste - wiggle the mouse a bit if the pasting seems to pause. Such are the mysteries of MS-Windows.) (10.394251 18.819084 6.825021 90.000000 106.437195 90.000000)
- Give the spacegroup (P 21/A) Note that this is a non-standard setting and may give problems later when viewing in "Default" Platon - in putting the connectivity out of whack. Also, different programs may handle the default connectivity differently.
- Give the wavelength. (1.52904)
- This is X-ray data (the codeword is 4)
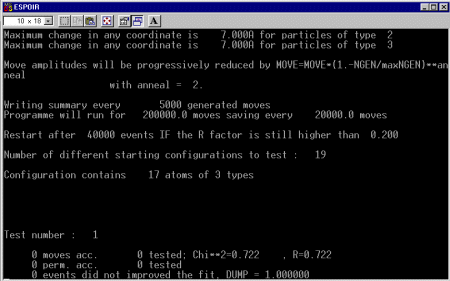
- There are 17 non-hydrogen atoms in this structure; thus enter 17
- There are 3 different atom types.
- There is one object (the freely moving atoms) - so enter 1
- When in doubt (especially if there is an overlap problem), fit on the regenerated pseudo-pattern: = 1
- Get the printouts.
- Give the U,V,W from the Le Bail fit. (0.00414 0.00042 0.00144)
- Give the number of points above for the pseudo powder pattern (I tend to say 4)
- When prompted, give the Atom Types one at a time. (S N C)
- We will not be providing bond contraints. (0 = no)
- Give maximum move (in Angstrom) for each atom type. Not sure of a good rule here - I guess you find out by trial and error(?), I just copied from the ESPOIR example site (7)
- Give the annealing law: go with the default: 2 (unless you have another option?)
- Give sigma : the data precision - go with the default: 1
- Give reject: I tend to go with 0.005 instead of the default of 0.001
- Give number of moves between prints (I tend to go for 5000)
- Give the maximum number of moves: Go with default of 200000
- Give the maximum number for saving: I tend to go with 20000
- If after nstart events, the R factor is larger than rmax, then restart Give nstart and rmax: (I tend to blindly go for the defaults: 40000 and 0.2) (so ESPOIR will reject any solution about 20% after 40,000 moves)
- I like to fit on "R", thus enter 2
- Go for the default number of Runs (10) - you may have to optimise this over time.
- IMPORTANT: Depending on the size and nuances of the structure, the number of
trials that it will take for ESPOIR to stumble on a solution could vary considerably. In the case
of Cemitidine, it has been found that 1 out of 50 trials may succeed - but you only find this out
the hard way. Thus take this into account in that if ESPOIR fails within a certain number of trials,
you may have to increase the number of trials; or consider changing some
of the options (and increasing the number of trials). Such is life with real-space structure
solution methods.
- Check the ESPOIR help area for advice in this regard: http://sdpd.univ-lemans.fr/sdpd/espoir/examples/index.html. Advice is available on the web about molecular organics include Cimetidine, Pyrene and 1-methylfluorene: http://sdpd.univ-lemans.fr/sdpd/espoir/examples/index.html#Cimetidine | http://sdpd.univ-lemans.fr/sdpd/espoir/examples/index.html#pyrene | http://sdpd.univ-lemans.fr/sdpd/espoir/examples/index.html#1-met
- IMPORTANT: Depending on the size and nuances of the structure, the number of
trials that it will take for ESPOIR to stumble on a solution could vary considerably. In the case
of Cemitidine, it has been found that 1 out of 50 trials may succeed - but you only find this out
the hard way. Thus take this into account in that if ESPOIR fails within a certain number of trials,
you may have to increase the number of trials; or consider changing some
of the options (and increasing the number of trials). Such is life with real-space structure
solution methods.
- For Object Number 1 (the only object in this example)
- We are using randomly generated atoms, and NPERM of 10 seems nice thus enter 1 10
- When prompted for number of Sulphur atoms: 1
- When prompted for number of Nitrogen atoms: 6
- When prompted for number of Carbon atoms: 10
- Enter a thermal value - ESPOIR suggests 3 for Organic.
- Using occupancy of 1 for all atoms: Enter 0
- Try a guess that all atoms are on general positions: Enter 0
- Enter any number (try 4?) to finish with PRESOIR