This example uses example Cimetidine data from the EXPO software
|
If EXPO gives problems, another option is to use fragment searching via single crystal (or Monte Carlo) methods.
However, the major problem you might initially encounter is how to generate the fragment. There are a number of programs that can take a 2D structure and try and convert it into a 3D structure. Refer: Starting Links to 2D to 3D Model Builders and Molecular Modelling Software In this tutorial, we will use the Web-based CORINA software (with Java building applet) to give us a 3D fragment. |
|
Go to the CORINA page
at http://www2.ccc.uni-erlangen.de/software/corina/free_struct.html
and scroll down until you see the data entry for the SMILES string.
|
|
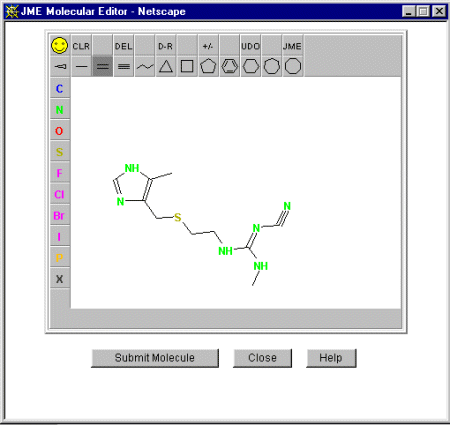
Select the Create Molecule button to bring up the JME Molecular Editor and
enter the fragment in 2D format. You may like to play with just entering a part of
the structure - or the entire structure - and see what the optimisation algoirthm
does. Here we have entered the entire molecule.
|
|
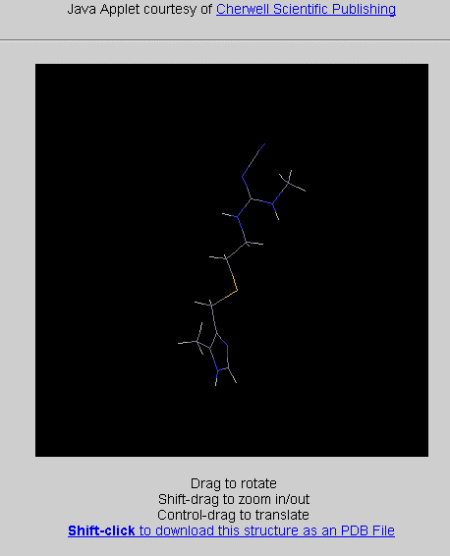
Now Submit the fragment to be minimized into a 3D conformation; and
viewed with the JAVA applet (smile string it uses being CNC(=NC#N)NCCSCc1nc[NH]c1C).
|
|
Save the now 3D fragment into PDB file ready to import into
various crystallographic software for possible structure solution.
Following is the PDB file. (Platon can directly import the PDB file as well)
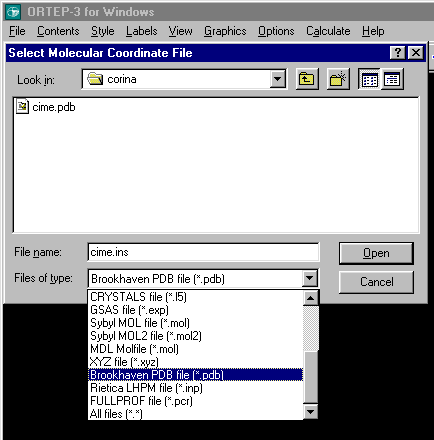
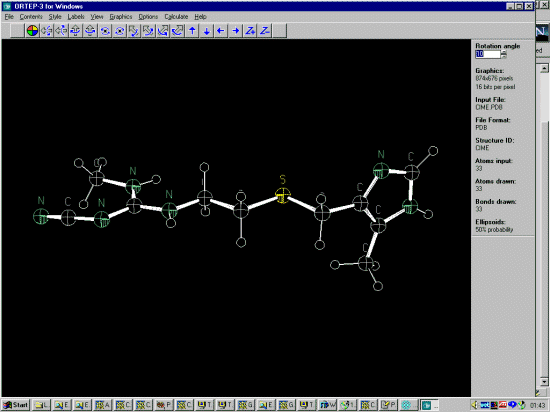
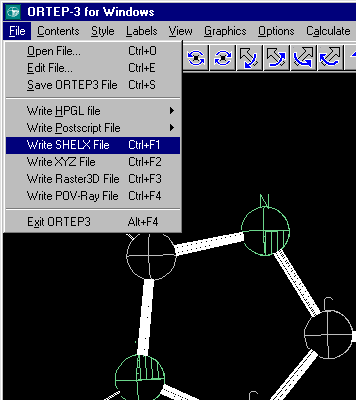
For instance, Ortep-3 by Louis Farrugia can import a PDB file and save it as a Shelx file in a ready to edit format.
Use File, Open File and the list box to the PDB format to import the PDB file.
Use File, Write SHELX File to output the file into Shelx format. (Though XYZ format is also possible)
|
PDF FileHEADER NONAME 16-Apr-00 NONE 1 TITLE NONE 2 AUTHOR SVR4 nobody uid NONE 3 REVDAT 1 16-Apr-00 0 NONE 4 ATOM 1 C 0 -5.282 -1.101 1.291 0.00 0.00 C+0 ATOM 2 N 0 -3.941 -0.740 0.825 0.00 0.00 N+0 ATOM 3 C 0 -3.792 0.071 -0.264 0.00 0.00 C+0 ATOM 4 N 0 -4.852 0.625 -0.826 0.00 0.00 N+0 ATOM 5 C 0 -6.069 0.503 -0.243 0.00 0.00 C+0 ATOM 6 N 0 -7.089 0.401 0.247 0.00 0.00 N+0 ATOM 7 N 0 -2.547 0.310 -0.773 0.00 0.00 N+0 ATOM 8 C 0 -1.362 -0.212 -0.086 0.00 0.00 C+0 ATOM 9 C 0 -0.101 0.297 -0.786 0.00 0.00 C+0 ATOM 10 S 0 1.365 -0.349 0.065 0.00 0.00 S+0 ATOM 11 C 0 2.672 0.395 -0.951 0.00 0.00 C+0 ATOM 12 C 0 4.019 -0.016 -0.416 0.00 0.00 C+0 ATOM 13 N 0 4.716 -1.101 -0.784 0.00 0.00 N+0 ATOM 14 C 0 5.836 -1.140 -0.115 0.00 0.00 C+0 ATOM 15 N 0 5.887 -0.071 0.707 0.00 0.00 N+0 ATOM 16 H 0 6.607 0.151 1.319 0.00 0.00 H+0 ATOM 17 C 0 4.737 0.645 0.512 0.00 0.00 C+0 ATOM 18 C 0 4.345 1.923 1.208 0.00 0.00 C+0 ATOM 19 H 0 -5.849 -1.532 0.467 0.00 0.00 H+0 ATOM 20 H 0 -5.202 -1.830 2.098 0.00 0.00 H+0 ATOM 21 H 0 -5.792 -0.210 1.656 0.00 0.00 H+0 ATOM 22 H 0 -3.159 -1.076 1.290 0.00 0.00 H+0 ATOM 23 H 0 -2.446 0.825 -1.588 0.00 0.00 H+0 ATOM 24 H 0 -1.365 0.127 0.950 0.00 0.00 H+0 ATOM 25 H 0 -1.378 -1.301 -0.112 0.00 0.00 H+0 ATOM 26 H 0 -0.099 -0.041 -1.822 0.00 0.00 H+0 ATOM 27 H 0 -0.086 1.387 -0.760 0.00 0.00 H+0 ATOM 28 H 0 2.570 0.053 -1.981 0.00 0.00 H+0 ATOM 29 H 0 2.583 1.481 -0.918 0.00 0.00 H+0 ATOM 30 H 0 6.596 -1.902 -0.205 0.00 0.00 H+0 ATOM 31 H 0 4.715 2.776 0.639 0.00 0.00 H+0 ATOM 32 H 0 3.259 1.979 1.282 0.00 0.00 H+0 ATOM 33 H 0 4.778 1.939 2.209 0.00 0.00 H+0 CONECT 1 2 19 20 21 NONE 38 CONECT 2 1 3 22 0 NONE 39 CONECT 3 2 4 7 0 NONE 40 CONECT 4 3 5 0 0 NONE 41 CONECT 5 4 6 0 0 NONE 42 CONECT 6 5 0 0 0 NONE 43 CONECT 7 3 8 23 0 NONE 44 CONECT 8 7 9 24 25 NONE 45 CONECT 9 8 10 26 27 NONE 46 CONECT 10 9 11 0 0 NONE 47 CONECT 11 10 12 28 29 NONE 48 CONECT 12 11 13 17 0 NONE 49 CONECT 13 12 14 0 0 NONE 50 CONECT 14 13 15 30 0 NONE 51 CONECT 15 14 16 17 0 NONE 52 CONECT 17 15 12 18 0 NONE 53 CONECT 18 17 31 32 33 NONE 54 END NONE 55
Shelx File
TITL
CELL 0.71073 10.0000 10.0000 10.0000 90.0000 90.0000 90.0000
ZERR 1 0.0010 0.0010 0.0010 0.0100 0.0100 0.0100
LATT 1
SFAC C N S H
UNIT 10 6 1 16
FVAR 1.0
H 4 -0.52020 -0.18300 0.20980 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
H 4 0.47780 0.19390 0.22090 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
H 4 -0.57920 -0.02100 0.16560 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
H 4 0.66070 0.01510 0.13190 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
C 1 -0.52820 -0.11010 0.12910 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
H 4 -0.31590 -0.10760 0.12900 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
C 1 0.43450 0.19230 0.12080 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
H 4 0.32590 0.19790 0.12820 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
N 2 0.58870 -0.00710 0.07070 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
N 2 -0.39410 -0.07400 0.08250 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
H 4 -0.58490 -0.15320 0.04670 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
H 4 -0.13650 0.01270 0.09500 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
N 2 -0.70890 0.04010 0.02470 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
C 1 0.47370 0.06450 0.05120 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
H 4 0.47150 0.27760 0.06390 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
H 4 0.65960 -0.19020 -0.02050 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
C 1 0.58360 -0.11400 -0.01150 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
S 3 0.13650 -0.03490 0.00650 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
C 1 -0.60690 0.05030 -0.02430 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
H 4 -0.13780 -0.13010 -0.01120 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
C 1 -0.13620 -0.02120 -0.00860 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
C 1 -0.37920 0.00710 -0.02640 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
C 1 0.40190 -0.00160 -0.04160 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
N 2 0.47160 -0.11010 -0.07840 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
N 2 -0.48520 0.06250 -0.08260 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
N 2 -0.25470 0.03100 -0.07730 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
C 1 -0.01010 0.02970 -0.07860 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
C 1 0.26720 0.03950 -0.09510 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
H 4 -0.00860 0.13870 -0.07600 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
H 4 0.25830 0.14810 -0.09180 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
H 4 -0.24460 0.08250 -0.15880 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
H 4 -0.00990 -0.00410 -0.18220 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
H 4 0.25700 0.00530 -0.19810 11.00000 0.03000 0.03000 =
0.03000 0.00000 0.00000 0.00000
HKLF 4
END
|