CCP14
Tutorials and Examples
CAOS - Single "Crystal Analysis Operating System" for Windows and UNIX
Example refinement of an Inorganic Phase Using CAOS
The CCP14 Homepage is at http://www.ccp14.ac.uk
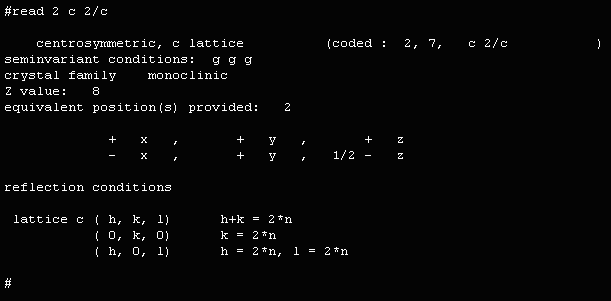
This example uses the raw hkl data used to determine the crystal
structure of Cs2TiSi6O15
as published in I.E. Grey, R.S. Roth, M.L. Balmer,
Journal of Solid State Chemistry, 131, 38-42 (1997).
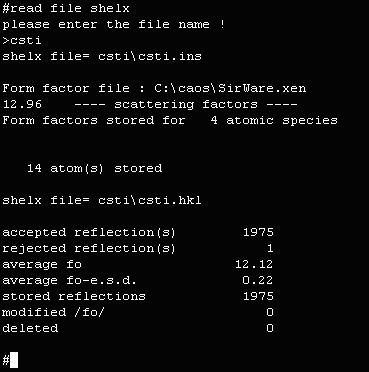
- Place the data in a subdirectory under the c:\caos directory (in this case c:\caos\csti). Each structure should
have it's own subdirectory.
- Run CAOS from either the Windows explorer or the CAOS Icon.

- This leads to the CAOS Screen that will prompt you to give the directory, and the
control filename.
- CAOS remembers the most recent structure it worked on so we will be over riding this.
When prompted for Current work file directory is:, enter CSTI and when
prompted to the binary file, type CSTI. If the binary file does not exist, CAOS
will create it. When prompted for the current input and output filename, just press enter.
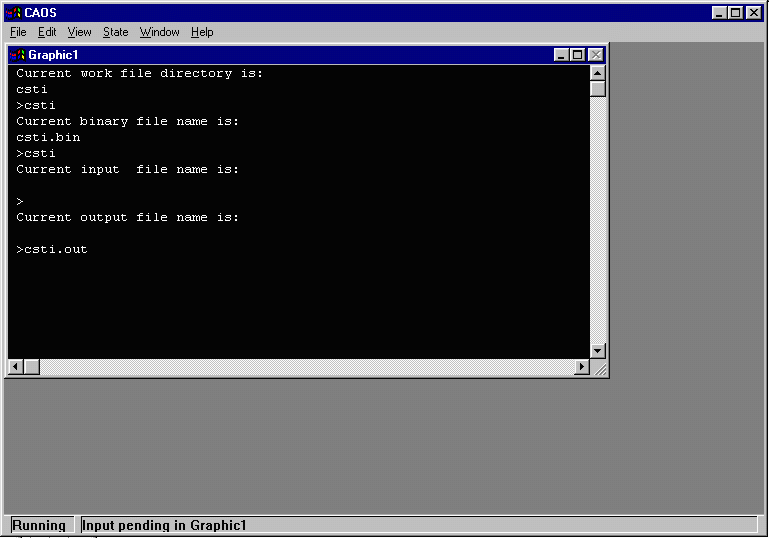
- As the binary file does not exist, CAOS will prompt whether you want to
crate it. Enter Y to create the binary file.
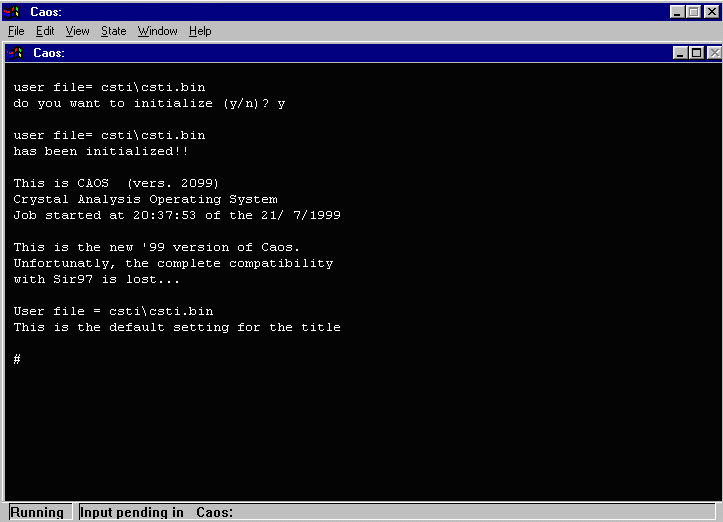
- At this point, maximise the screen text window so that the command prompt
is always visible on the screen (otherwise it might be one line below the screen)
- To do some cycles of refinement (by default the imported atoms
will refine positions and isotropic thermals), at the # prompt type LSQ then [enter]
and if you want to refine to convergence, type ## then [enter].
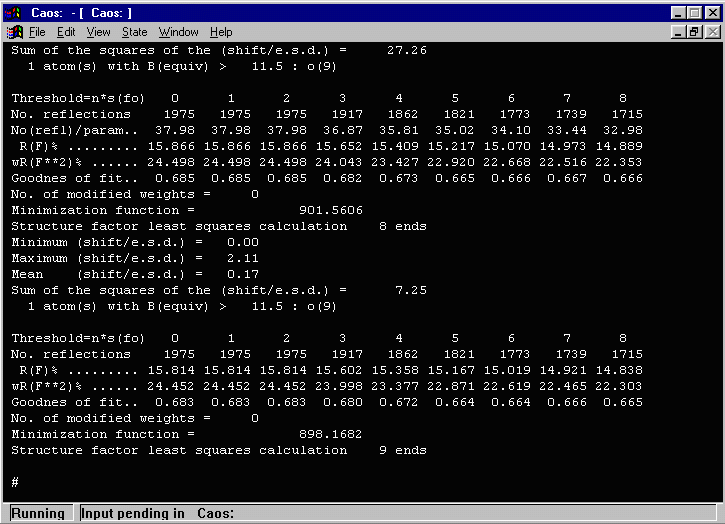
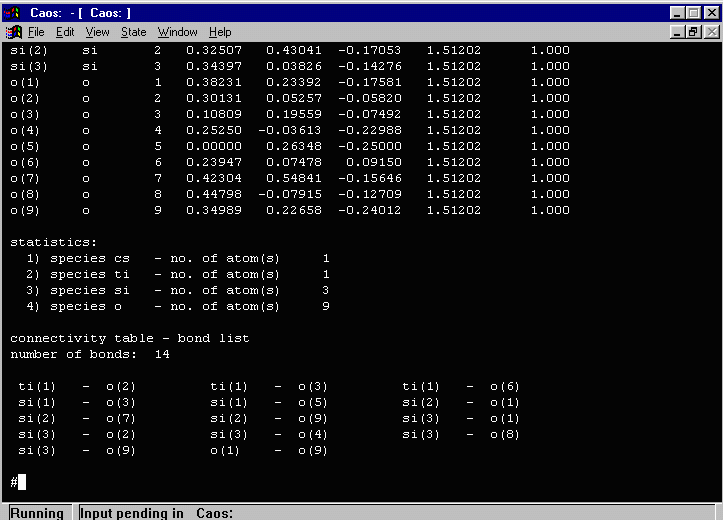
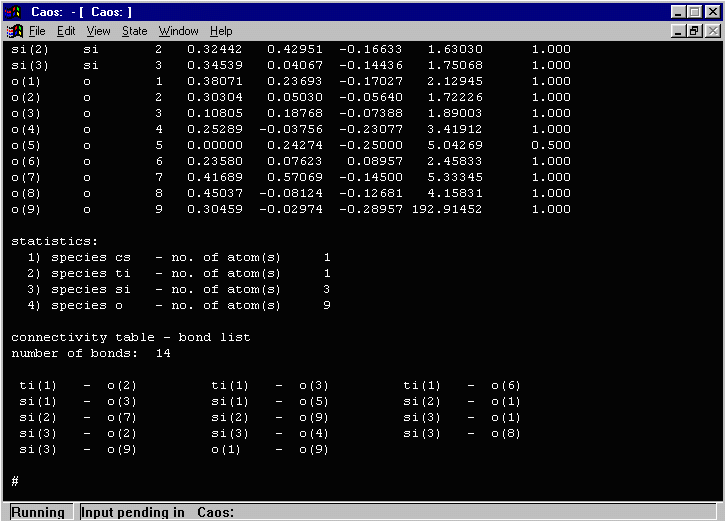
- In the above output, CAOS warns that one atom (O(9)) has an isotropic
thermal greater than 11.5. Doing a LIST 5 (or a DUMP FILE to dump all the
list contents to a file for evaluation) shows that the O(9) thermal (of 192!) is most likely
a spurious atom and should be deleted.
- While CAOS has its own structure viewer in-built into the program, it is possible
to use external structure viewers that can input CIF files. To output a CIF file, at
the # prompt, type CIF and when prompted, enter a file name. Then import the CIF
file into a structure viewer such as Loius Farrugia's Ortep-3 or GUI WinSTRUPLO.
If CAOS refuses to create a CIF file stating that "no list of type 14 stored";
the way around this is at the # prompt type FOURIER [enter] and ## [enter]
and this will populate LIST 14 such that you can now routinely create CIF files.
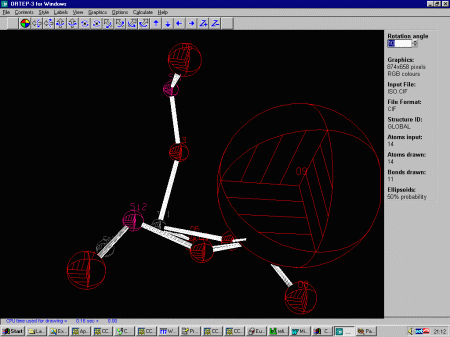
- Importing the outputted CIF file into Ortep-3 graphically shows
that o(9) is definitely got a problem.
- To delete the offending Oxygen atom:
- At the # prompt type update atoms [enter]
- at the > prompt type delete o(9) [enter]
- at the > prompt type end [enter]
- To again to least squares until convergence
- At the # prompt type LSQ [enter]
- at the > prompt type ## [enter]
- Remember that you can write out a CIF file at
any time and view the structure with Ortep-3 or GUI WinSTRUPLO
to keep a graphical track of the structure. If you write to the
identical file name, you can do a reload of the structure to
view the update.
- To now set the heavy Cs and Ti atoms refinding anisotropic:
- At the # prompt type update atoms [enter]
- at the > prompt type aniso cs ti [enter]
- at the > prompt type end [enter]
- To again apply least squares until convergence
- At the # prompt type LSQ [enter]
- at the > prompt type ## [enter]
- Remember that you can write out a CIF file at
any time and view the structure with Ortep-3 or GUI WinSTRUPLO
to keep a graphical track of the structure. If you write to the
identical file name, you can do a reload of the structure to
view the update.
- To now set the Si atoms refinding anisotropic:
- At the # prompt type update atoms [enter]
- at the > prompt type aniso si [enter]
- at the > prompt type end [enter]
- To again apply least squares until convergence
- At the # prompt type LSQ [enter]
- at the > prompt type ## [enter]
- Remember that you can write out a CIF file at
any time and view the structure with Ortep-3 or GUI WinSTRUPLO
to keep a graphical track of the structure. If you write to the
identical file name, you can do a reload of the structure to
view the update.
- To now set the O atoms refinding anisotropic:
- At the # prompt type update atoms [enter]
- at the > prompt type aniso o [enter]
- at the > prompt type end [enter]
- To again apply least squares until convergence
- At the # prompt type LSQ [enter]
- at the > prompt type ## [enter]
- Remember that you can write out a CIF file at
any time and view the structure with Ortep-3 or GUI WinSTRUPLO
to keep a graphical track of the structure. If you write to the
identical file name, you can do a reload of the structure to
view the update.