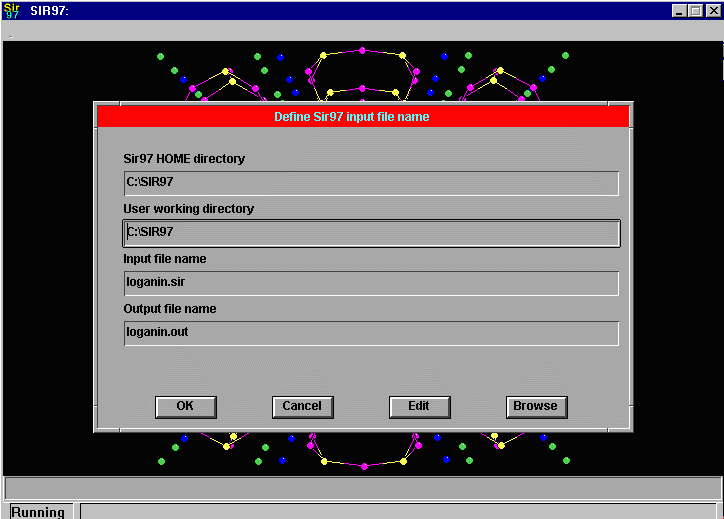
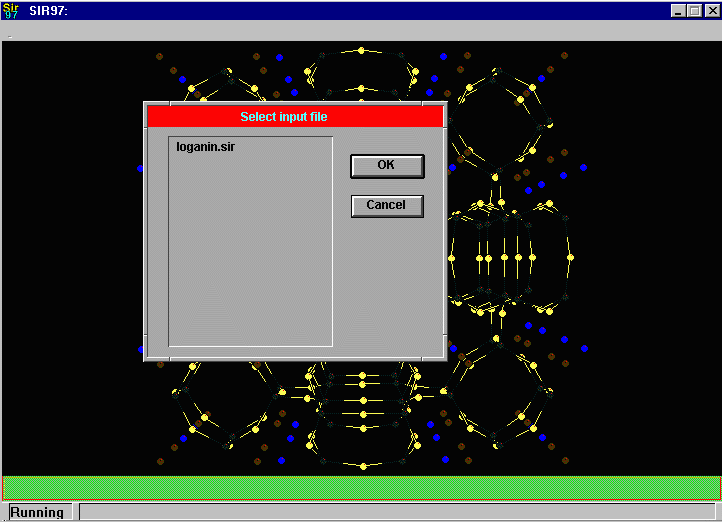
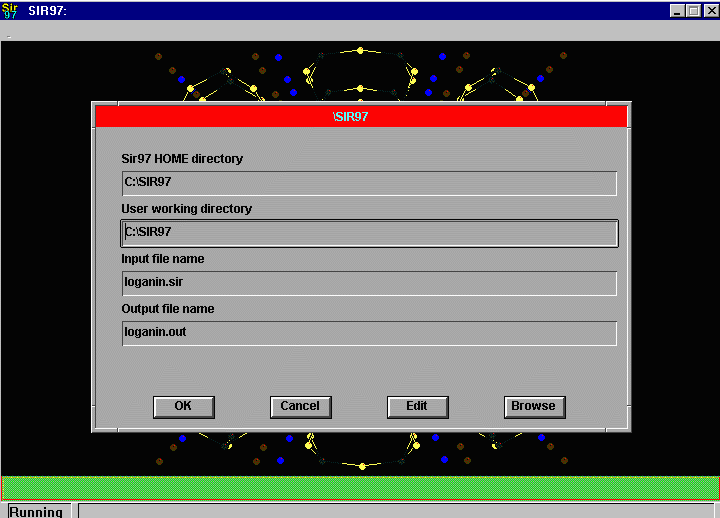
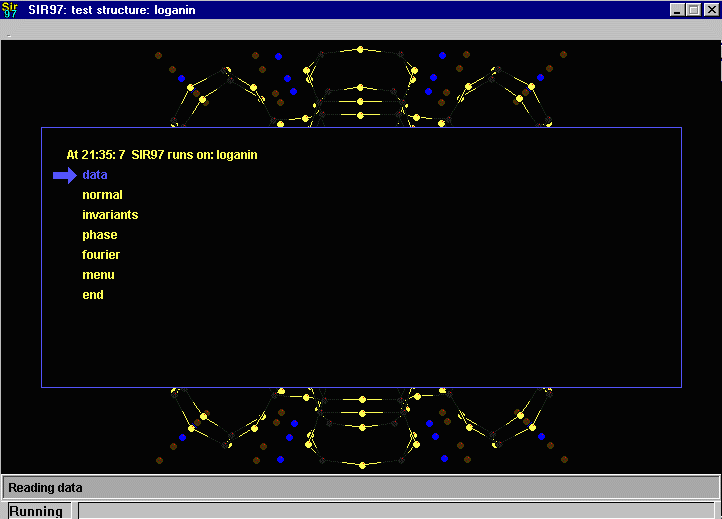
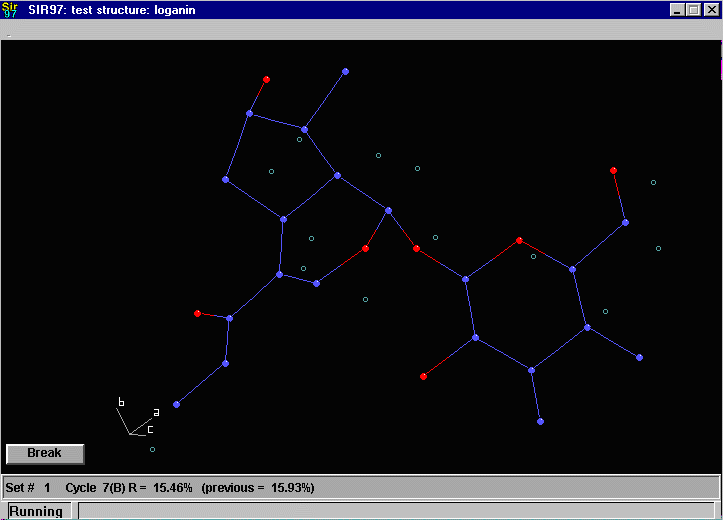
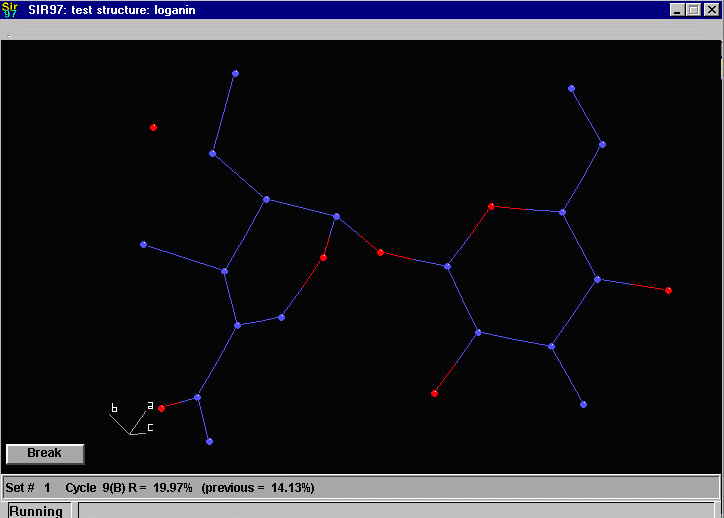
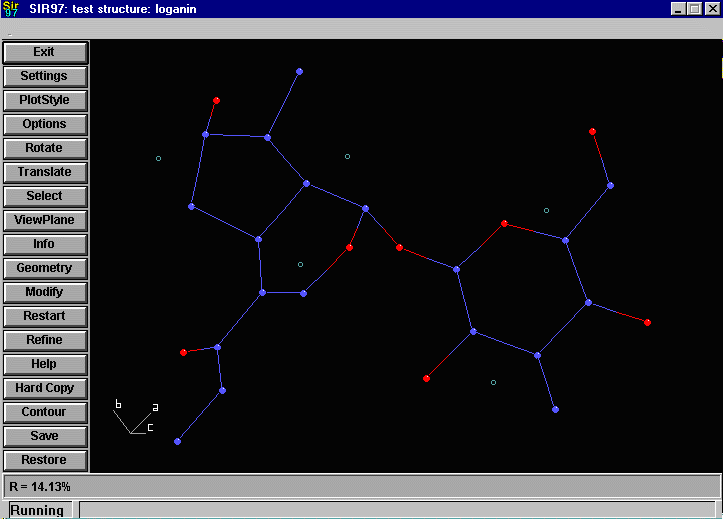
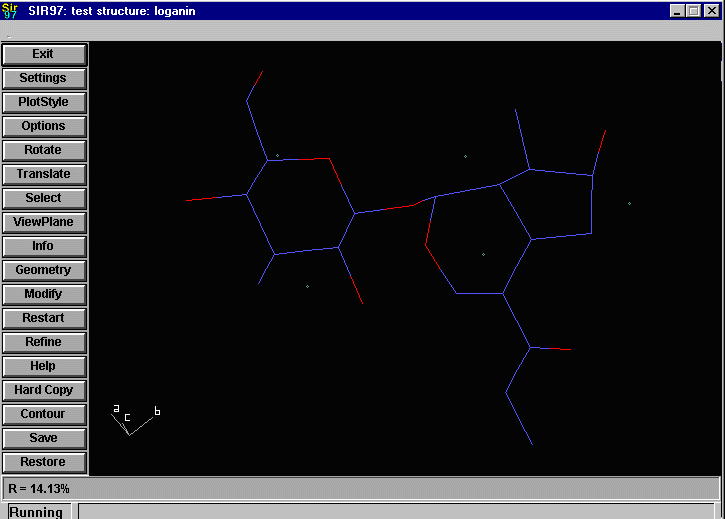
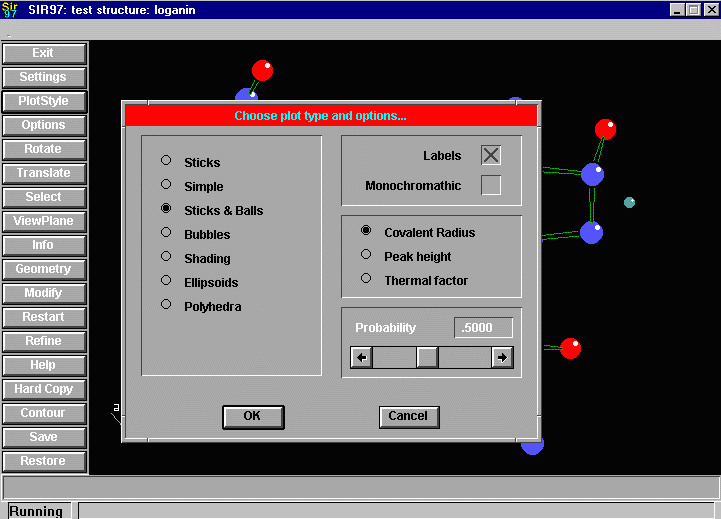
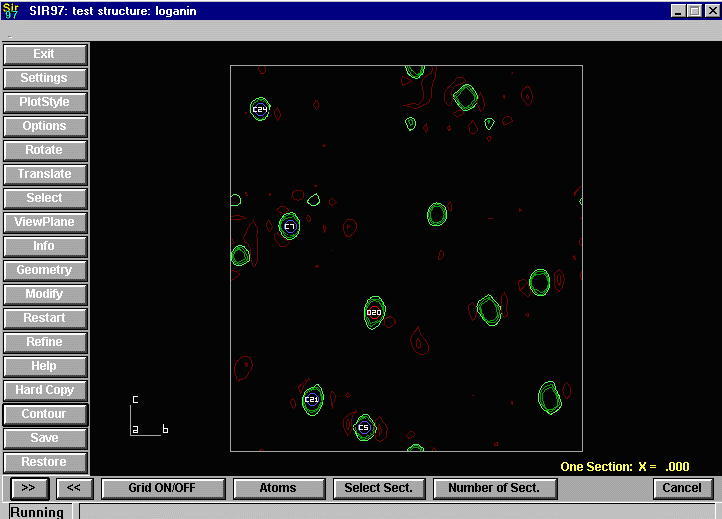
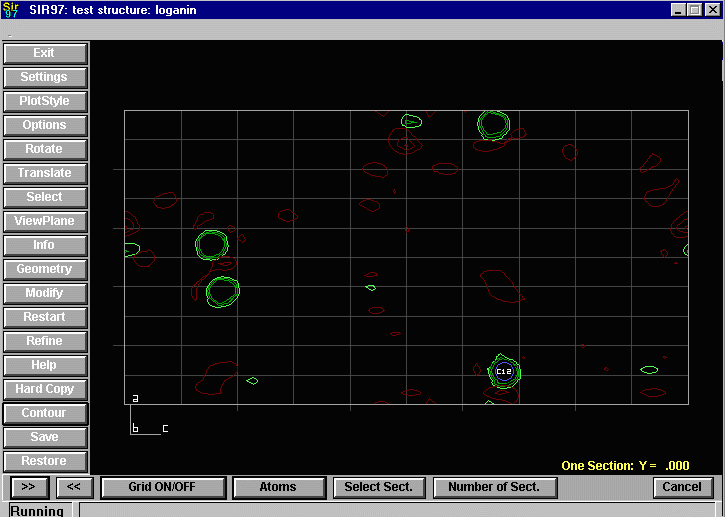
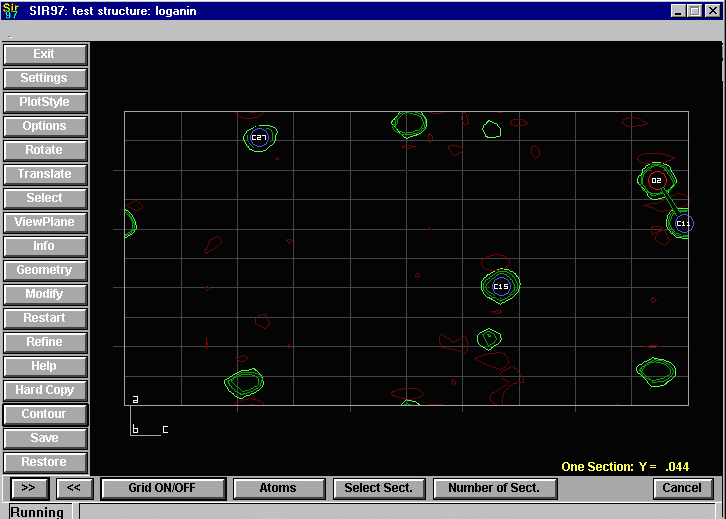
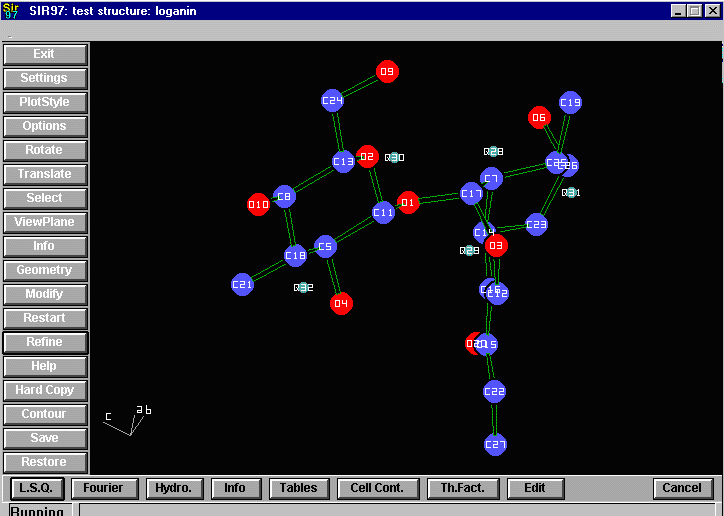
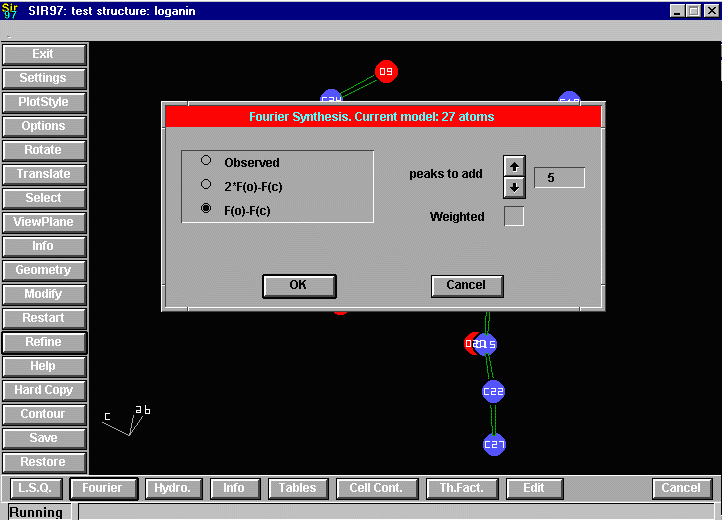
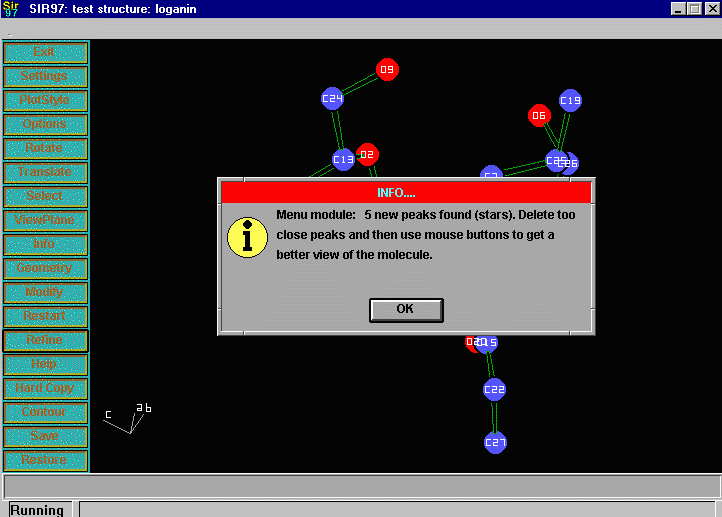
Sir2002 is now released and has solved structures with up to 2000 atoms in the asymmetric unit.
Refer to the main page for more information
Sir direct methods structure solution software
can be spawned transparently from the following single crystal suites
(freely available for students and academics) :
Note: a feature of Sir97 that may not be obvious to people solving
routine solvable structures (or who have mainly used Sir92) is if Sir97 states
that an adequate solution has not been found after Fourier cycling (R factor too high);
it will automatically continue until it either solves the structure, or exhausts
its strategy options.