|
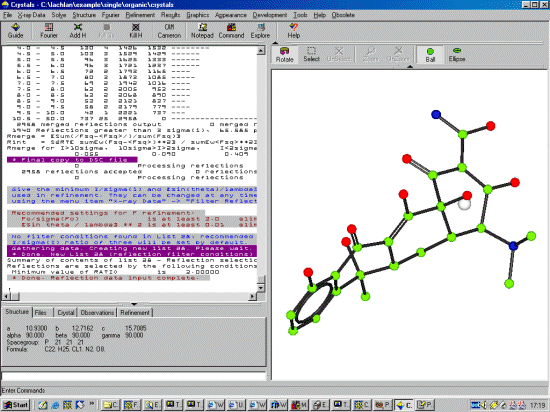
Run Crystals, select File, New and go to the directory
where the HKL data and Dirdif solution is located.
|
|
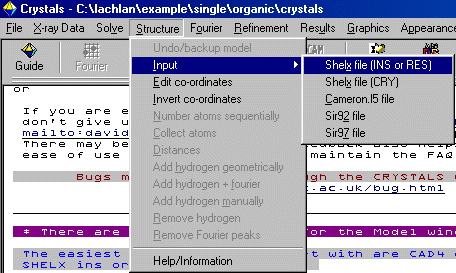
Select Structure, Input, Shelx File (INS or RES) to import the Dirdif solution which is in a Shelx format. Crystals will prompt for the
space group [P 21 21 21] (which is not part of the Shelx format), then will continue in importing the information from the Shelx file. If
Crystals has a problem with a kosher Shelx file, it could be you inserted the Space Group file in the wrong format.
If Crystals prompts if you want to change Q atoms to carbons, select Yes. If prompted to view the structure in Cameron, select No. If prompted to renumber the atoms, Yes or No are both fine choices in this case. Crystals will at this point try and assign bond types to the relevant parts of the structure (aromatic, double bond, triple bond).
|
|
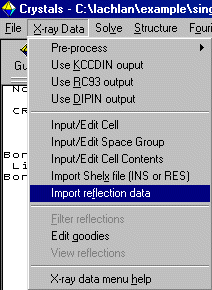
As Crystals can do DLS refinement, reflection data is an option you need to
explicitely set. Thus to insert the Fsquare reflection data,
select X-ray Data, Reflection Data to import the HKL Intensity file.
In this case, keep the cell and spacegroup information.
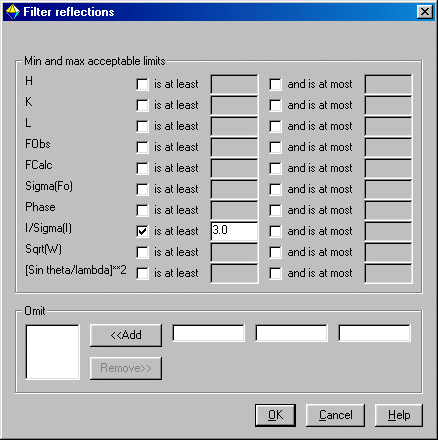
When prompted about Friedel Pairs, read the information and make a decision. When given the Filter Reflections menu, Crystals likes to get rid of the less than 3 sigma data (as detectors can often over determine the weak intensity data)
|
|
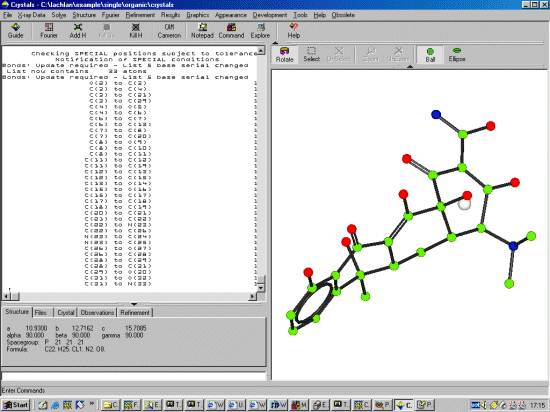
In the above case, DIRDIF has got all the atoms correct. However, If the
connectivity was not as it should be, this would be a good
time to relabel the atoms if you know the connectivity. If atoms do
get mislabelled, it should be possible to also figure this out later on
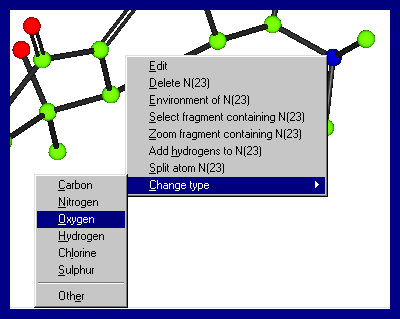
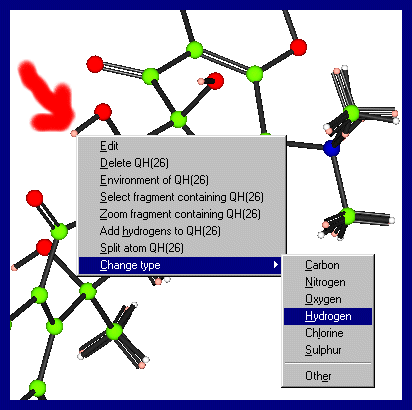
from the bondlengths/thermal values. Clicking Right Mouse Button over the structure and atoms will give you various options, including atom selection, deselection, zooming and unzooming To change an atom type, put the mouse over the atom of interest, click right mouse button (which brings up a menu) and go into Change type and select the atom of interest.
|
|
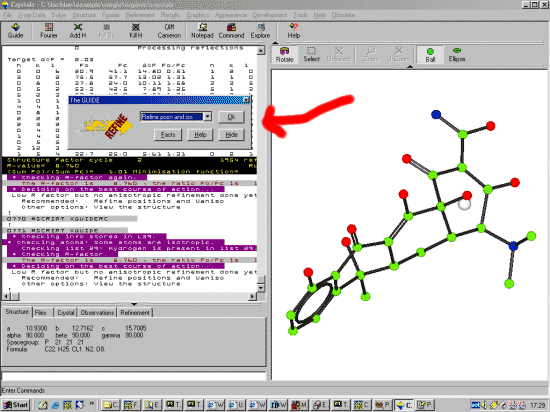
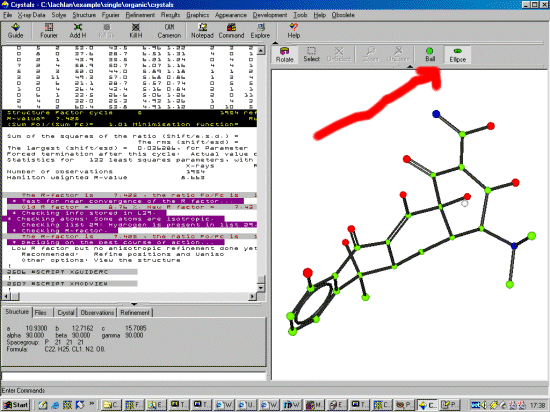
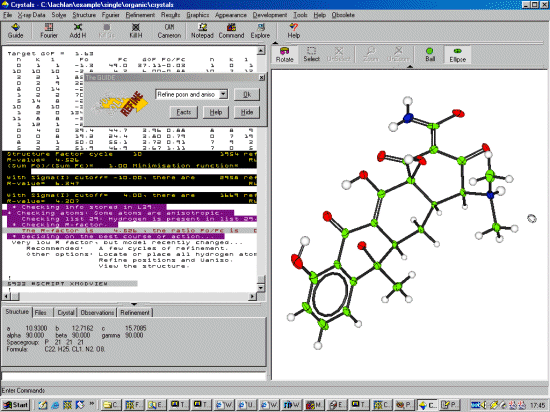
To start the Crystals Guide, select the Top Left Guide Icon. Or via the menu,
Refinement, Guided Automatic to start the refinement. Crystals
will do a few cycles of refinement, then the Supervisor
Dialog will then come up to guide you through the refinement.
|
|
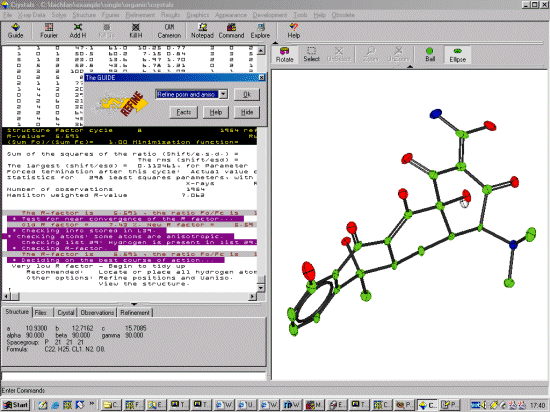
Because DIRDIF has done such a good job at solving and building up
the structure, (and the R factor is very good). Crystals
recommends going straight to adding hydrogens. However it would
be best to go through ISO, then ANSIO refinement before
choosing this option. Thus select "Refine Posn and Iso" and
click OK to start refining the structure.
|
|
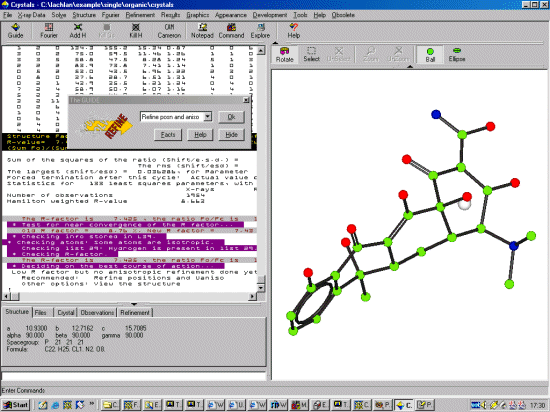
Crystals now recommends that you continue on with anisotropic refinement
but before doing this, it would be best to check out the
thermals and other nuances. Thus click on the Top Right Ellipse Icon.
|
|
Select Refinement, Guided Automatic to restart the guided refinement. The
Supervisor Dialog will then again come up to guide you
through the refinement, in this case now refine the atomic positions and
anisotropic thermals. Click OK to make this happen.
Thermal Ellipsoids will be displayed in the model windows
|
|
Crystals now recommends that you do hydrogen placement.
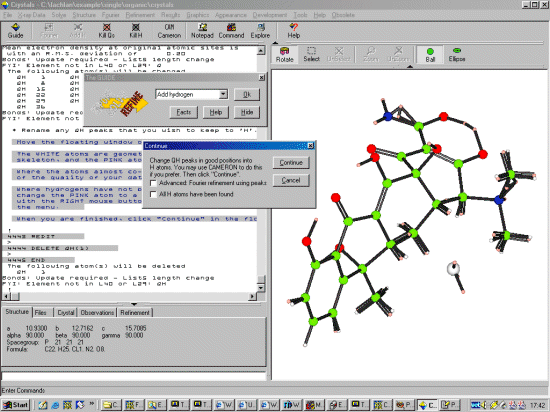
Select OK to do Hydrogen Placement after which Crystals will overlap calculated hydrogen positions with peaks found in the difference map.
When Crystals prompts with a Continue button, move this out of the way and examine the placed and found hydrogens. At present, Crystals only places hydrogens on the Carbons. If the placed and found hydrogens on the carbons match up, this is saying good things about the quality of the data to be able to see hydrogens. You will notice that it looks like the hydrogens have also be found for the Nitrogens and Oxygens.
Thus you can rename the Q peaks off the Oxygens and Nitrogens using your mouse. If you find, you cannot change a Q peak to a hydrogen, it could be the previous renaming has not finished yet (depending on the speed of your computer) Then accept the placed hydrogens off the Carbons by bringing back the Continue, and accept the GEOMETRIC (carbon) hydrogens
|
|
After doing some more rounds of refinement (ride the hydrogens if prompted for this);
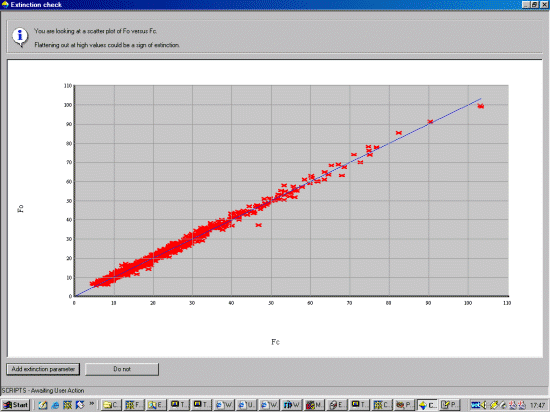
now it would be a good time to Check Extinction. So continue on with this.
Crystals will bring up an Fobs vs Fcalc plot. Read the information and select
whether you wish to refine extinction based on this. In this case, extinction is
not selected.
|
|
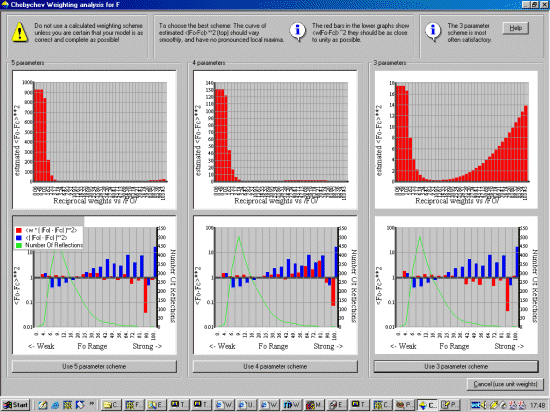
Now optimise the weighting scheme. Thus continue on and select "optimal"
for weighting scheme determination. Based on the graphs and information
Crystals gives you, select the weighting scheme parameters that seem most
appropriate. In this case a 3 parameter scheme.
|
|
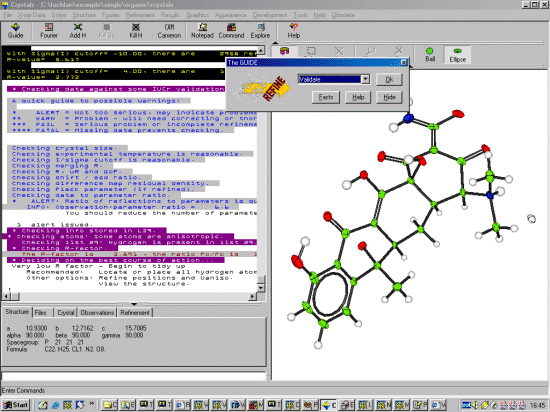
After doing some more cycles of refinement,
now let Crystals Validate what has been done so far to see if
there are is anything that is possibly dodgy.
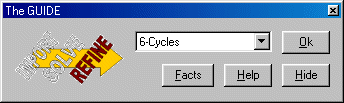
If crystals finds that the refinement has not fully converged (it often does at this point) do some unconditional cycles of refinement via the Guide using the 6-cycles option.
|