|
For this section, refer to the following part of the Crystals manual for more
complete options and explanations:
http://www.xtl.ox.ac.uk/crystalsmanual-9.html#9.3
The following is not all that realistic a structure to use but gives an idea of how DLS in Crystals can be performed. It should also be noted that it is expected that future versions of Crystals will allow the following to be done via the GUI (Graphical User Interface) as well. Also refer to Randomly Perturbing Atomic Positions in Crystals. This can be relevant for successfully performing DLS and refinement involving pseudo-symmetry. |
|
Import the structure you are interested in looking at. In this case, an inorganic Lithium Titanate
which is in Shelx format. The Molecule viewer in crystals is not optimised for polymeric
inorganics so don't be too worried if things look a bit ugly.
|
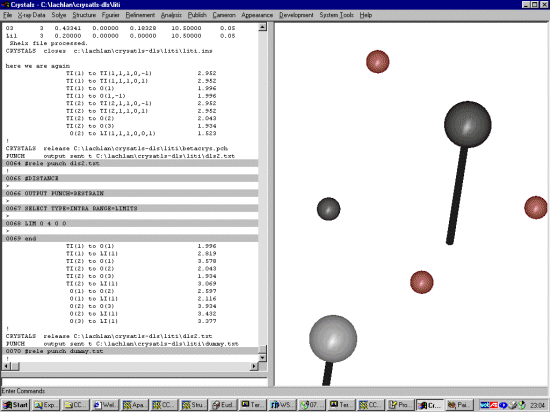
Generating Bond Distances and Bond AnglesYou may have to play with the limits to get the coverage of bonds that you are interested. DLS template restraints can be created by (the following no spaces between the # and command):
For inorganics, it may be relevant to generate bonds for all symmetry generated atoms, so the following may be a better option to make use of (depending on the aims of your DLS calculations).
|
Just Bond DistancesYou may have to play with the limits to get the coverage of bonds that you are interested. DLS template restraints can be created by (the following no spaces between the # and command):
|
|
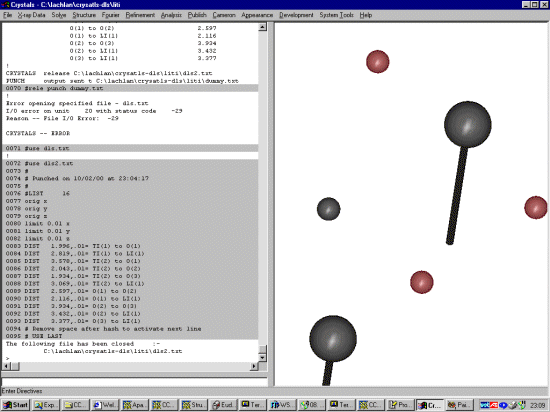
Edit the dls.txt (or whatever you have called this file) and change the distances/angle restraint with whatever ESD you consider appropriate to what you wish to use. The default ESD's for bond lengths are 0.01 Angstrom and for bond angles 1 degree. You may have to play with the restraint ESDs to get the desired level of flexibility. Also fix the origin and use limits to stop the refinement sending the co-ordinates to outer-space. Thus edit the dls.txt file in your favourite text editor and add: orig x orig y orig z limit 0.01 x limit 0.01 y limit 0.01 z
The file might now looking something like this: # # Punched on 10/02/00 at 23:04:17 # #LIST 16 orig x orig y orig z limit 0.01 x limit 0.01 y limit 0.01 z DIST 1.996,.01= TI(1) to O(1) DIST 2.819,.01= TI(1) to LI(1) DIST 3.578,.01= TI(2) to O(1) DIST 2.043,.01= TI(2) to O(2) DIST 1.934,.01= TI(2) to O(3) DIST 3.069,.01= TI(2) to LI(1) DIST 2.597,.01= O(1) to O(2) DIST 2.116,.01= O(1) to LI(1) DIST 3.934,.01= O(2) to O(3) DIST 3.432,.01= O(2) to LI(1) DIST 3.377,.01= O(3) to LI(1) # Remove space after hash to activate next line # USE LAST |
|
Once you are happy with the restraints in the dls.txt file, instruct
Crystals to use the above list of restraints and commands type
#use dls.txt end
|
| In the present version of Crystals, it is necessary to give Crystals a dummy HKL file - which we then later tall to ignore. Thus go into X-ray Data, Shelx file (INS and RES) |
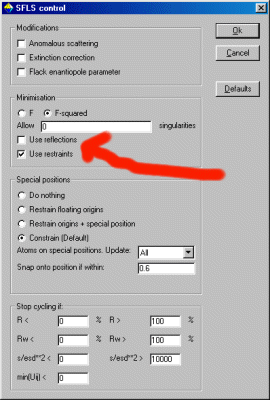
Performing DLS refinement: telling Crystals that you do not want to refine on HKLsIn the latest GUI version of Crystals, you can use the Refinement, F or F2 and advanced options GUI to instruct Crystals not to use reflections. Just declick the Use Reflections checkbox but make sure the Use Restraints checkbox is "on".
You can also use the following command line method to tell Crystals that you "do not want" to refine on any HKLs (answer the questions this script queries you on).
#script inlist23 When prompted with: Do you wish to use LIST 6 reflections? (keyword [YES]) Type NO From David Watkin: "But since crystals has no way of predicting what distortion of the structure you would like to try, you have to modify them by hand. HOWEVER, all the symmetry related bonds and angles are listed, and since this technique is usually used for high symmetry materials, this is a great saving. In addition to the distance and angle restraints, you can also add in any/all of the other features. Its a great way for saying 'what if?' and trying a new SG or something." From David Watkin: "LIST 23 is the refinement control list (NOT the parameter definition list - that is LIST 12, nor the parameters themselves, that is LIST 5!). Here you can say if you want to use reflections, restraints or both. If you use neither, CRYSTALS issues a warning!" |
| You also modify restraints using the GUI Interface by selecting the required atoms, then selecting the restraint using the Right Mouse Button to bring up a list of GUI options. |
|
Crystals is instructed to refine the atomic positions of the restrained atoms using the following:
#list 12 block x's end |
|
To refine, use either from the script command line:
\SFLS REFINE END Or from the GUI menu, select Refinement, Least Squares |
|
To go back one refinement step you can refer to Multiple UNDO in Crystals - restoring old structure models in Crystals or use:
#disk reset 5 0 -1 end |