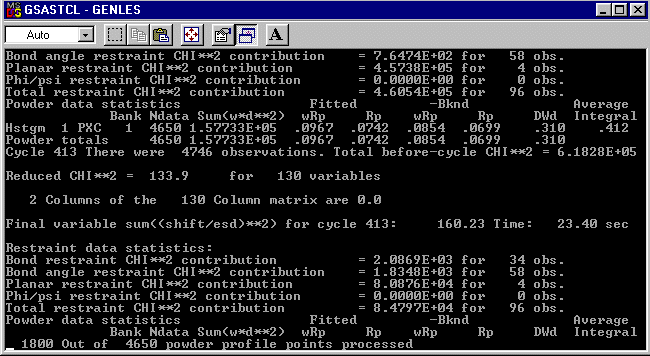
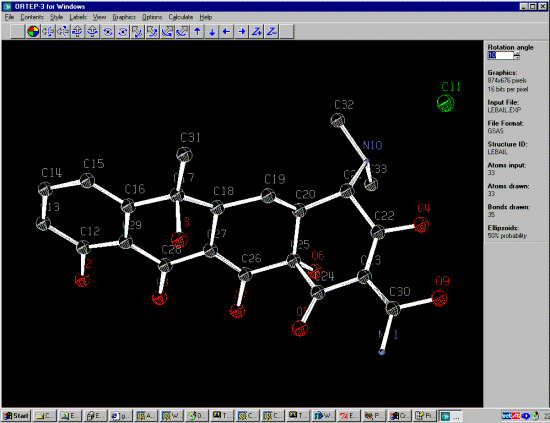
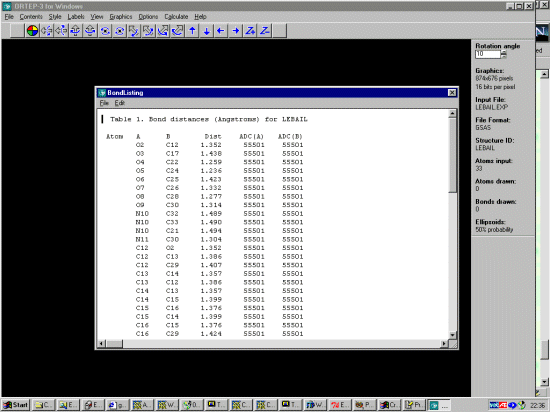
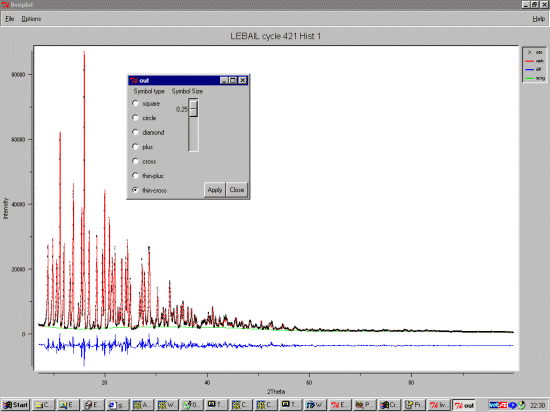
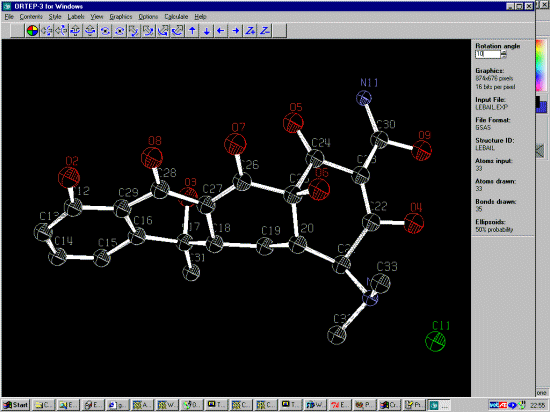
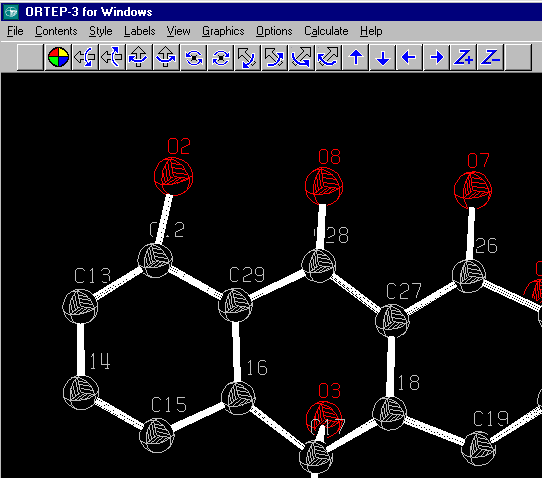
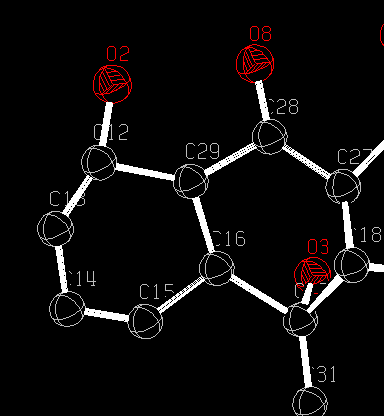
DIST 1.352,.01= O(2) TO C(12)
DIST 1.432,.01= O(3) TO C(17)
DIST 1.259,.01= O(4) TO C(22)
DIST 1.234,.01= O(5) TO C(24)
DIST 1.422,.01= O(6) TO C(25)
DIST 1.330,.01= O(7) TO C(26)
DIST 1.276,.01= O(8) TO C(28)
DIST 1.313,.01= O(9) TO C(30)
DIST 1.494,.01= N(10) TO C(21)
DIST 1.490,.01= N(10) TO C(32)
DIST 1.491,.01= N(10) TO C(33)
DIST 1.301,.01= N(11) TO C(30)
DIST 1.386,.01= C(12) TO C(13)
DIST 1.405,.01= C(12) TO C(29)
DIST 1.357,.01= C(13) TO C(14)
DIST 1.399,.01= C(14) TO C(15)
DIST 1.375,.01= C(15) TO C(16)
DIST 1.534,.01= C(16) TO C(17)
DIST 1.422,.01= C(16) TO C(29)
DIST 1.540,.01= C(17) TO C(18)
DIST 1.516,.01= C(17) TO C(31)
DIST 1.526,.01= C(18) TO C(19)
DIST 1.512,.01= C(18) TO C(27)
DIST 1.534,.01= C(19) TO C(20)
DIST 1.538,.01= C(20) TO C(21)
DIST 1.522,.01= C(20) TO C(25)
DIST 1.522,.01= C(21) TO C(22)
DIST 1.403,.01= C(22) TO C(23)
DIST 1.427,.01= C(23) TO C(24)
DIST 1.435,.01= C(23) TO C(30)
DIST 1.552,.01= C(24) TO C(25)
DIST 1.508,.01= C(25) TO C(26)
DIST 1.354,.01= C(26) TO C(27)
DIST 1.445,.01= C(27) TO C(28)
DIST 1.451,.01= C(28) TO C(29)
ANGL 111,20= C(21) TO N(10) TO C(32)
ANGL 115,20= C(21) TO N(10) TO C(33)
ANGL 111,20= C(32) TO N(10) TO C(33)
ANGL 118,20= O(2) TO C(12) TO C(13)
ANGL 121,20= O(2) TO C(12) TO C(29)
ANGL 121,20= C(13) TO C(12) TO C(29)
ANGL 119,20= C(12) TO C(13) TO C(14)
ANGL 122,20= C(13) TO C(14) TO C(15)
ANGL 120,20= C(14) TO C(15) TO C(16)
ANGL 123,20= C(15) TO C(16) TO C(17)
ANGL 119,20= C(15) TO C(16) TO C(29)
ANGL 117,20= C(17) TO C(16) TO C(29)
ANGL 108,20= O(3) TO C(17) TO C(16)
ANGL 105,20= O(3) TO C(17) TO C(18)
ANGL 108,20= C(16) TO C(17) TO C(18)
ANGL 110,20= O(3) TO C(17) TO C(31)
ANGL 113,20= C(16) TO C(17) TO C(31)
ANGL 112,20= C(18) TO C(17) TO C(31)
ANGL 113,20= C(17) TO C(18) TO C(19)
ANGL 109,20= C(17) TO C(18) TO C(27)
ANGL 111,20= C(19) TO C(18) TO C(27)
ANGL 112,20= C(18) TO C(19) TO C(20)
ANGL 110,20= C(19) TO C(20) TO C(21)
ANGL 110,20= C(19) TO C(20) TO C(25)
ANGL 113,20= C(21) TO C(20) TO C(25)
ANGL 115,20= N(10) TO C(21) TO C(20)
ANGL 110,20= N(10) TO C(21) TO C(22)
ANGL 117,20= C(20) TO C(21) TO C(22)
ANGL 117,20= O(4) TO C(22) TO C(21)
ANGL 124,20= O(4) TO C(22) TO C(23)
ANGL 118,20= C(21) TO C(22) TO C(23)
ANGL 121,20= C(22) TO C(23) TO C(24)
ANGL 117,20= C(22) TO C(23) TO C(30)
ANGL 121,20= C(24) TO C(23) TO C(30)
ANGL 125,20= O(5) TO C(24) TO C(23)
ANGL 120,20= O(5) TO C(24) TO C(25)
ANGL 115,20= C(23) TO C(24) TO C(25)
ANGL 109,20= O(6) TO C(25) TO C(20)
ANGL 105,20= O(6) TO C(25) TO C(24)
ANGL 110,20= C(20) TO C(25) TO C(24)
ANGL 111,20= O(6) TO C(25) TO C(26)
ANGL 111,20= C(20) TO C(25) TO C(26)
ANGL 110,20= C(24) TO C(25) TO C(26)
ANGL 113,20= O(7) TO C(26) TO C(25)
ANGL 123,20= O(7) TO C(26) TO C(27)
ANGL 124,20= C(25) TO C(26) TO C(27)
ANGL 123,20= C(18) TO C(27) TO C(26)
ANGL 118,20= C(18) TO C(27) TO C(28)
ANGL 119,20= C(26) TO C(27) TO C(28)
ANGL 121,20= O(8) TO C(28) TO C(27)
ANGL 120,20= O(8) TO C(28) TO C(29)
ANGL 119,20= C(27) TO C(28) TO C(29)
ANGL 119,20= C(12) TO C(29) TO C(16)
ANGL 121,20= C(12) TO C(29) TO C(28)
ANGL 120,20= C(16) TO C(29) TO C(28)
ANGL 118,20= O(9) TO C(30) TO N(11)
ANGL 120,20= O(9) TO C(30) TO C(23)
ANGL 122,20= N(11) TO C(30) TO C(23)
PLANAR 0.010000 O(2) C(12) C(13) C(14) C(15) C(16) C(29)