 SYSTEM-S SYSTEM-S
SYSTEM-S SYSTEM-S
 PLATON HOMEPAGE
PLATON HOMEPAGE
Look here for an alternative System-S tutorial by Lachlan Cranswick (CCP14).
The external tools (SHELXS97, SHELXL97, DIRDIF96/99, SIR97, CRUNCH)
are used 'as supplied' by their respective authors and should
be obtained from the sources given below. They are NOT provided as part
of this distribution. S will NOT work when no SHELX-97 executables are
found in the PATH.
The NQA-mode obviously works only in the case of (relatively) trouble free
structures (i.e. no disorder, twinning and similar specialist issues).
External:
Note: CRUNCH should run in the foreground (as opposed to the
default:background)
Build-in:
If not, environment parameters should be set:
Example for setting PLAEXE in the (t)csh-shell:
setenv PLAEXE /mnt/spea/bin/platon
when the platon executable is in /mnt/spea/bin
Note: no spacegroup information required. Data/instruction
lines other than the above are ignored.
The CELL should be consistent with that of the reflection file.
sdemo.hkl contains a standard shelx HKLF 4 style dataset
S can now be run in the auto-mode (the No-Questions-Asked mode)
with the keyboard instruction:
s sdemo.ins nqa
After some time (during which the space group is determined,
the structure is solved by direct methods (SHELXS) and refined (SHELXL)
including H-atoms) the result of the analysis is shown as a rotating
molecule.
The rotation can be stopped by clicking in the window.
S can be terminated by typing 'END'.
Alternatively, S can be started in the 'guided' mode via:
s sdemo.ins
An interactive sequence is set up in response.
User input routinely involves 'hitting-the-return-key' when
the suggested material within [] is o.k (or clicking on
ACCEPT-DEFAULT in the menu bar).
An 'END' instruction terminates S.
Remarks:
Directory 's' will be created automatically in the current directory
unless there exists already such a directory in '~USER'.
A second example (C13 H24 N2 Pd), illustrating a structure determined
automatically by heavy atom methods (DIRDIF99), can be run with
sdemo1.ins and sdemo1.hkl.
Start the structure determination with s sdemo.ins
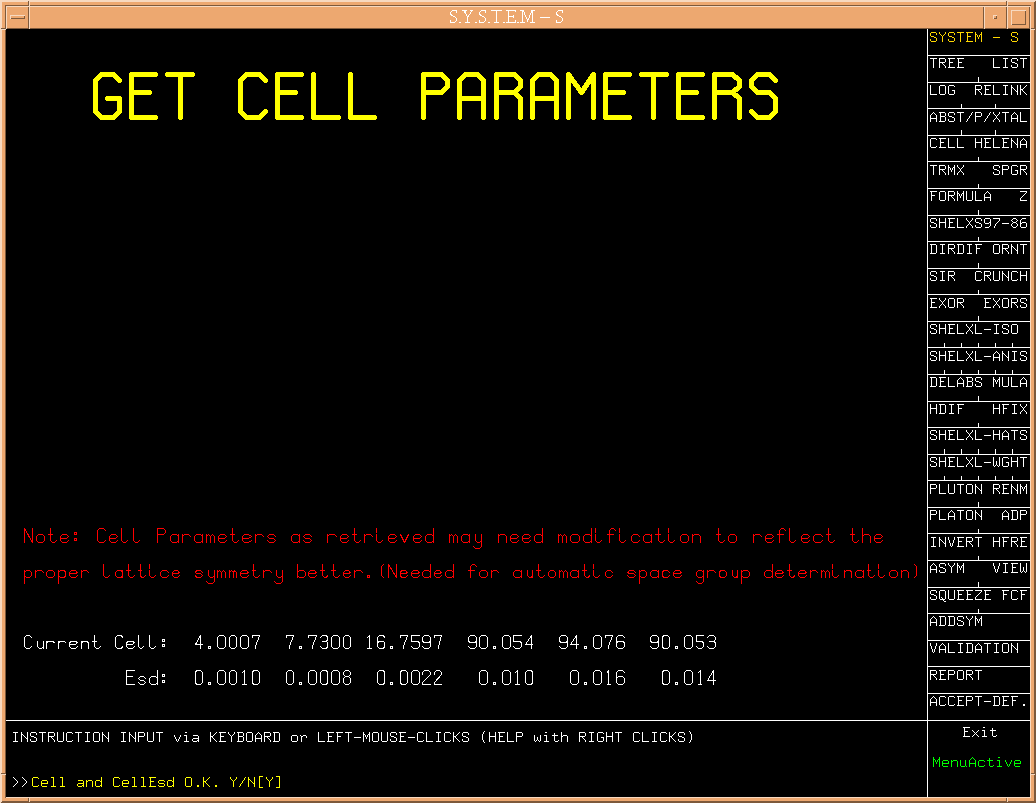
The s-shell prompt should look like s[CELL]
(Fig.1)
Hitting the return key (or clicking on ACCEPT-DEF)
will bring up the section of S that
establishes the cell dimensions and associated esd's. In the current
case we just acknowledge (with a return) the correctness of the values found in
sdemo.ins. When desired, cell data may be changed at this point.
(Fig.1a)
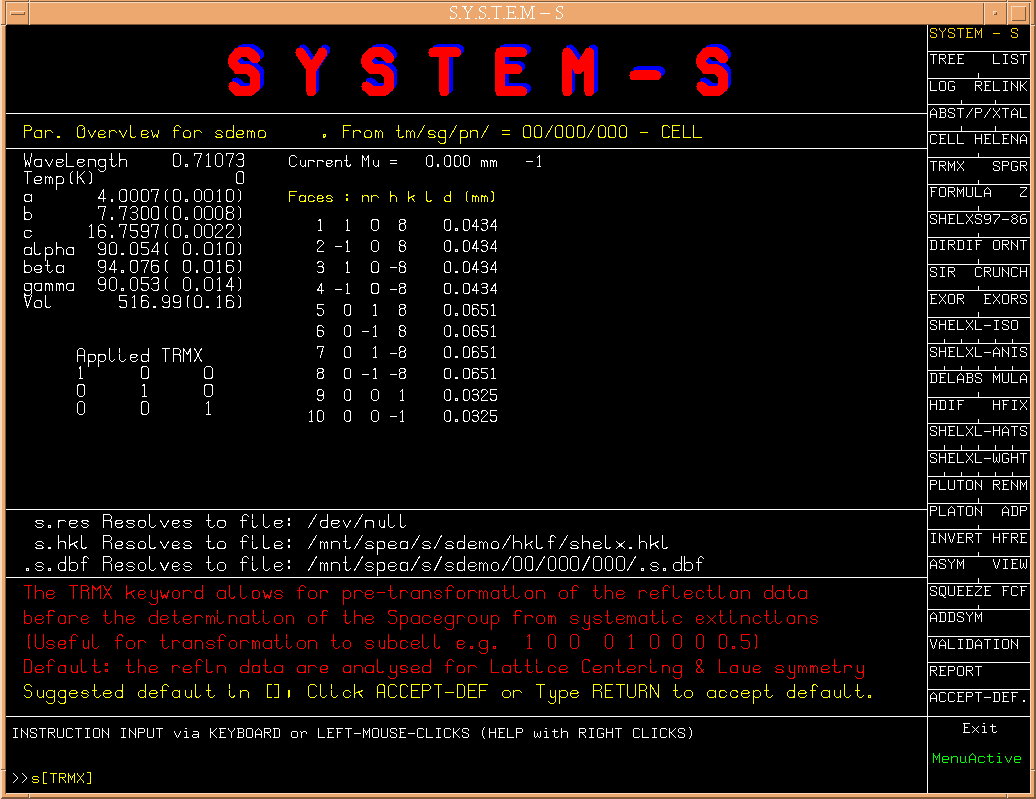
The next suggested logical step, i.e s[TRMX],
(Fig.2)
is the determination
of the lattice type and Laue group with associated transformation matrix.
Hitting the return will bring up a number of options.
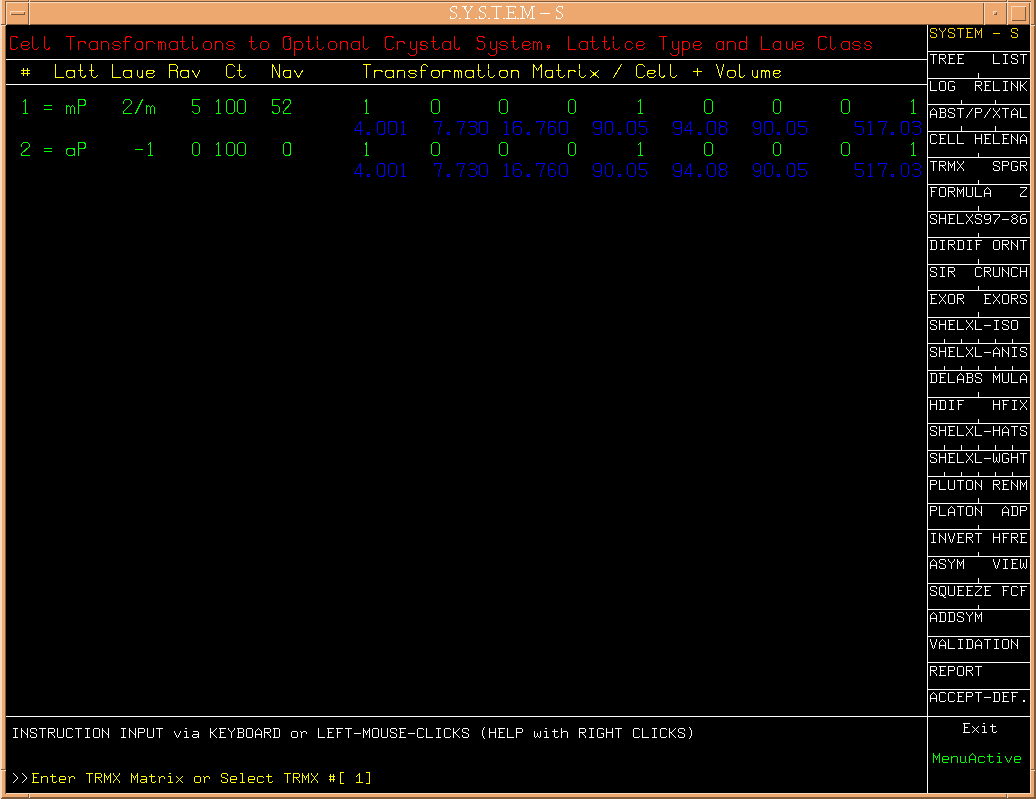
(Fig.3)
The suggested
choise (#1) is accepted again with a RETURN. The second option (#2)
could be attempted lateron when #1 doesn't lead to results.
(Fig.3a)
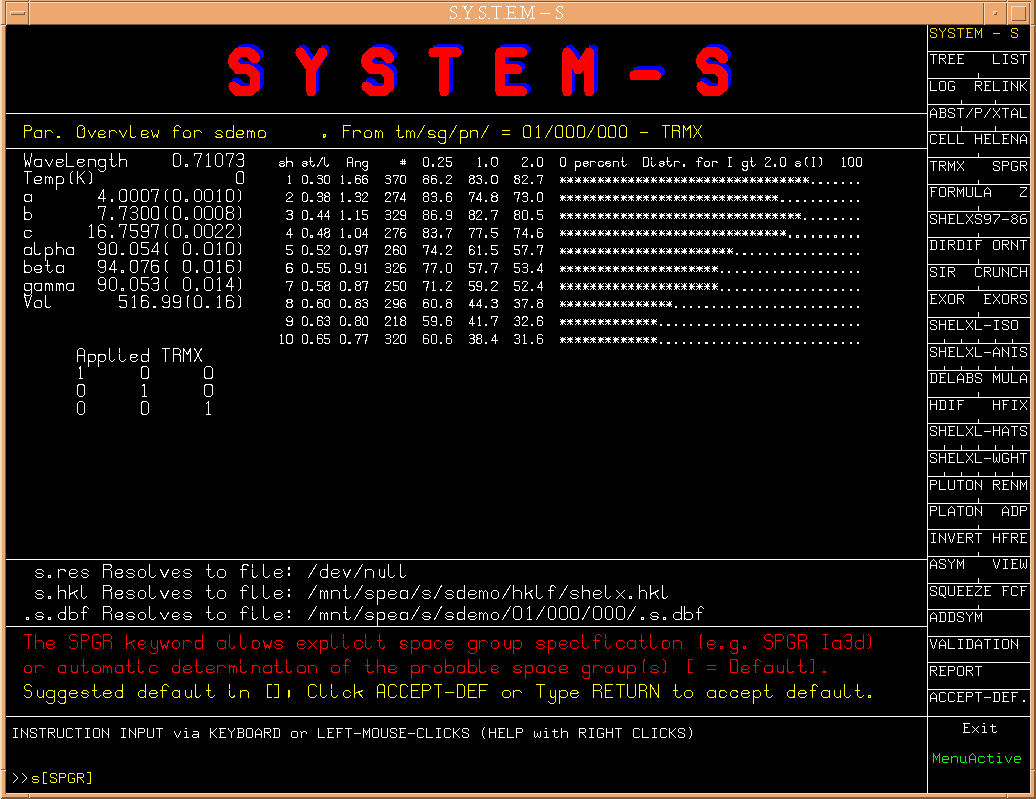
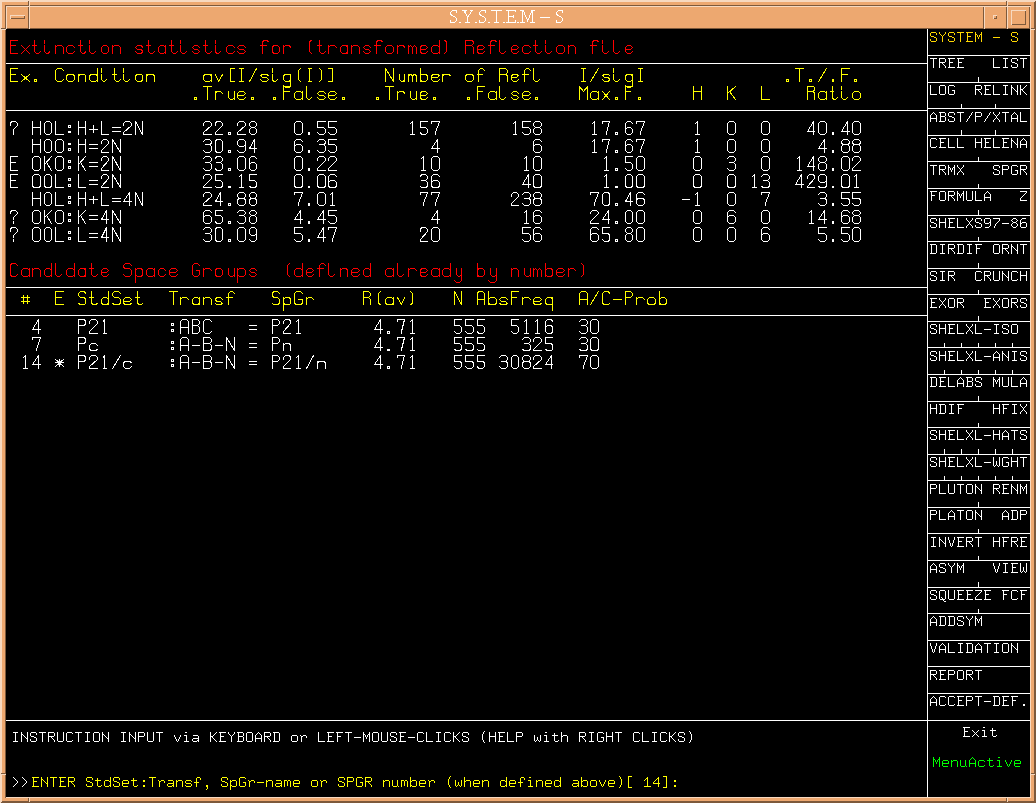
The next step s[SPGR] brings up the section of the space group
specification. A number of choises,
(Fig.4)
based on the observed systematic
extinctions, is suggested, along with an a-priori choise (#14).
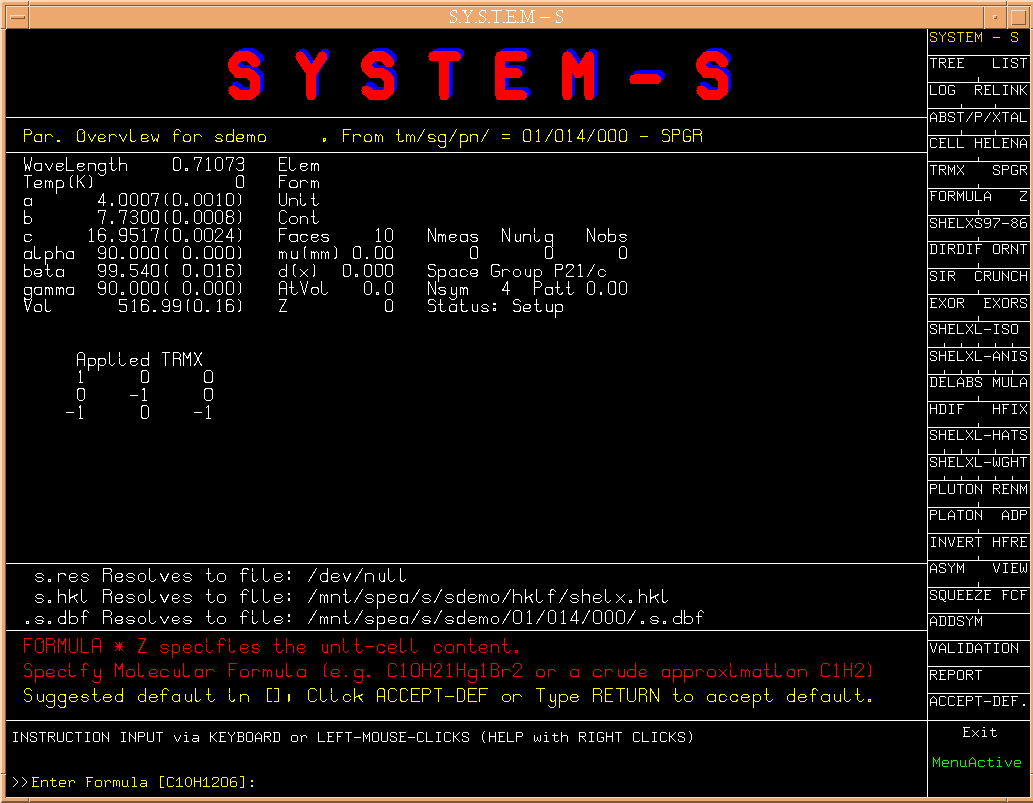
The next step s[FORMULA] brings up the section to specify
the cell content. In our case, the formula given in sdemo.ins is
suggested.
(Fig.5)
However any other specification is possible. Here we accept
the suggestion.
This brings up [Z]. Hitting RETURN will generate a suggested
value for Z (i.e. 2). Any other reasonable value may be entered here.
In our case we just hit RETURN again.
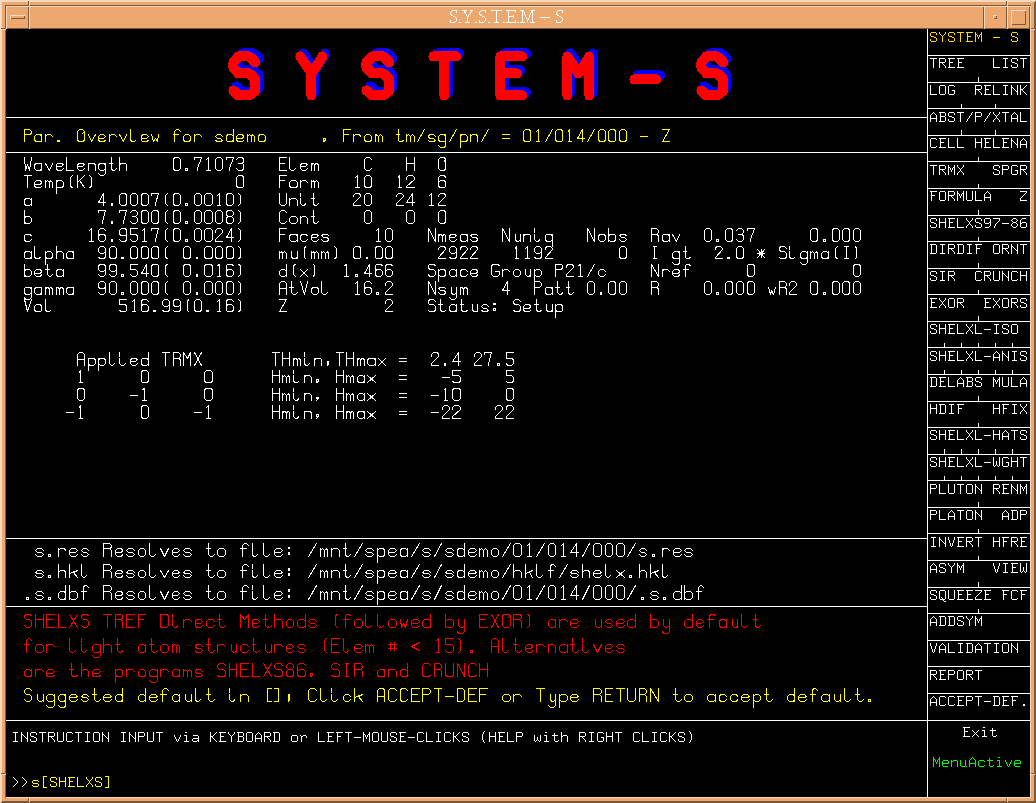
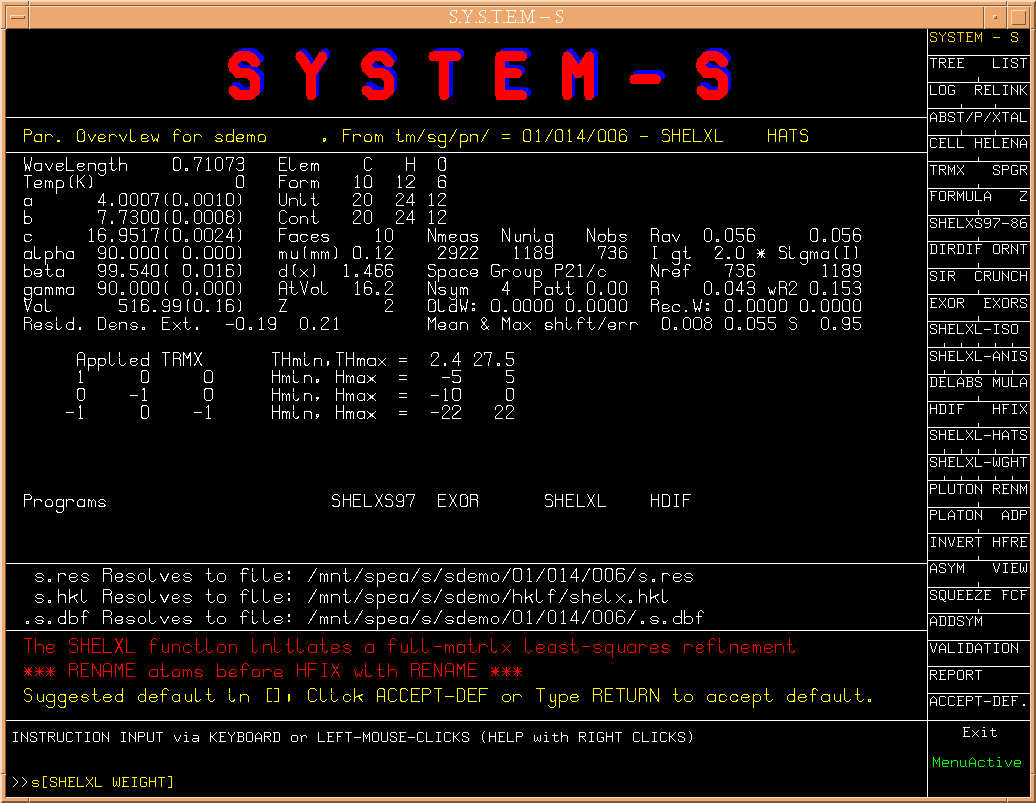
This brings us to the core of a structure determination, i.e. the
phase determination. S suggests to run SHELXS for this.
(Fig.6)
Alternatively,
the older SHELXS86 (as opposed to SHELXS97), SIR or CRUNCH could be
attempted when available on the machine. Here we again take the default
choise.
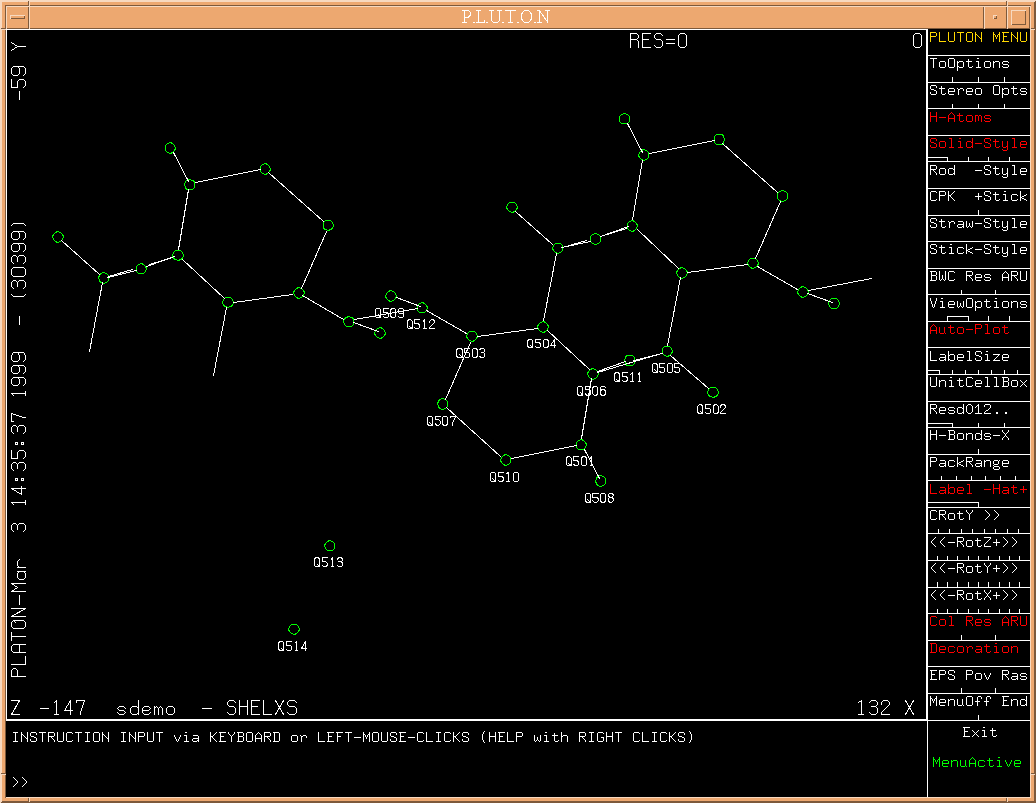
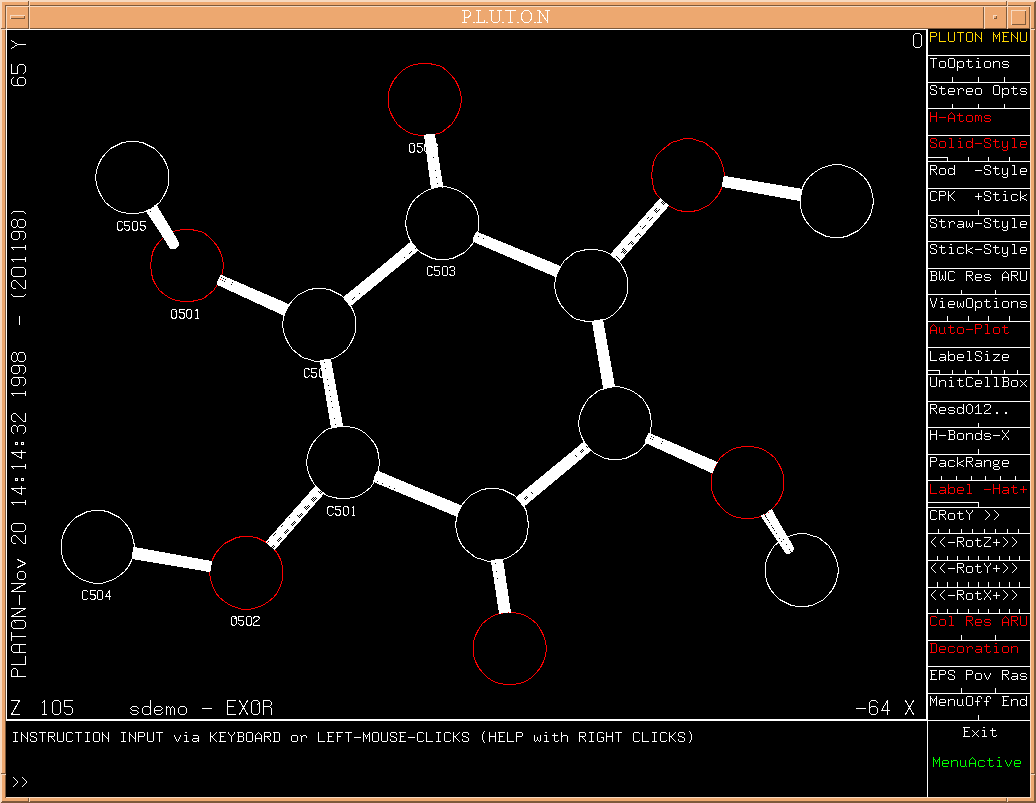
The result of the SHELXS/TREF calculation is now shown for inspection
with PLUTON.
(Fig.7)
PLUTON can be terminated by clicking on EXIT.
The list of atoms generated with SHELXS obviously needs some 'cleanup'.
This can be done with a procedure called EXOR (short for exorcise).
Indeed, all noise peaks have been removed and element types correctly
assigned as shown in
(Fig.8)
If not, as may be the case with more problematic structures,
PLUTON may be used to RENAME atoms to their desired labels
and atom types for those mis-assigned by the automatic procedure.
Also, remaining ghosts may be removed from the current model
(stored in s.res; and optionally to be inspected by clicking
on the LstRES Menu-button) with PLUTON.
On the termination of PLUTON, S suggests isotropic refinement as the
next step:
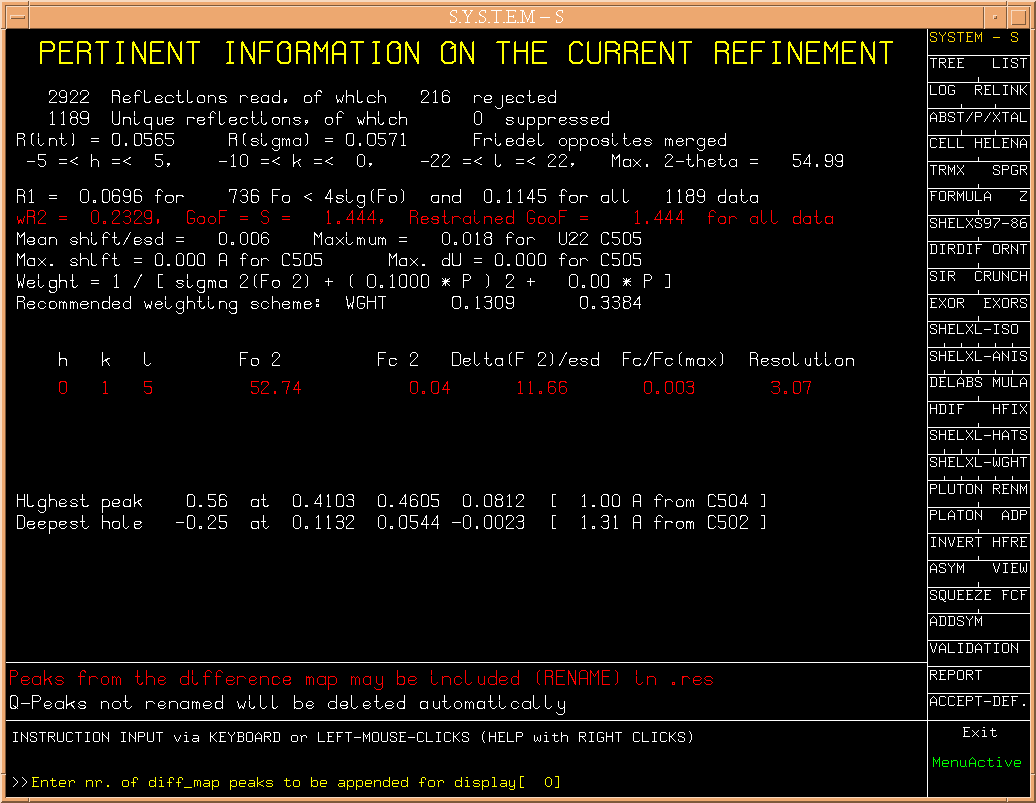
At the end of the refinement
a difference map is calculated from which the highest peaks can be
appended to the parameter file to be inspected with PLUTON. In the
current case, no significant residual density is found. Hit RETURN.
This step is followed by anisotropic refinement indicated with
s[SHELXL ANISO].
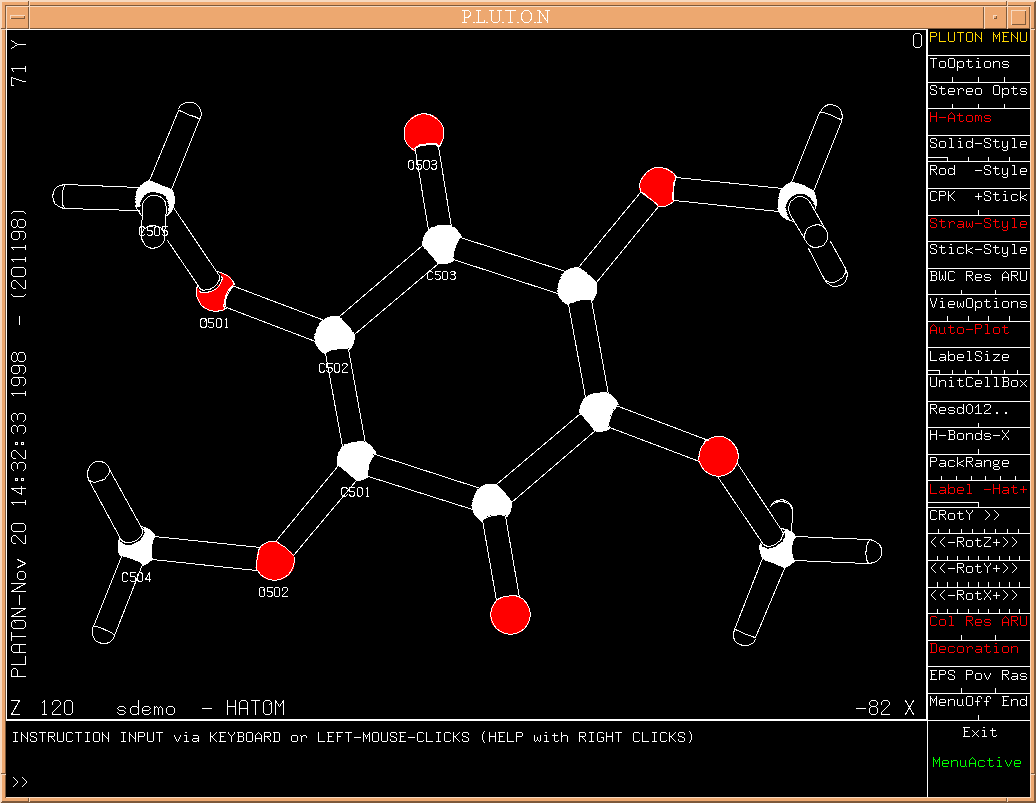
In the next step it is suggested to find H-atom positions in a
difference fourier map which is effected by hitting the RETURN key
on s[HATOMS]. The result is again shown in a PLUTON plot.
(Fig.9)
The atom list may be edited in this stage.
Intermediate SHELXL refinement results and warning display look like
(Fig.9a)
In the next step, H-atoms are included in the refinement.
The final step involves a weighted refinement.
(Fig.10)
This step can be
repeated until complete convergence.
Terminate with END.
The results of the current refinement are in ~USER/s/sdemo/tm/sg/shelxl.
Each project (structure) has its own subdirectory tree starting
with the name of the structure (e.g. sdemo).
The top-level directory for a structure is indicated as 'level-0'
Sub-directories of a level-0 directory include level-1 trees
(one for each lattice type that is attempted).
Sub-directories of a level-1 directory include level-2 trees
(one for each space group attempted to solve the structure in).
Level-2 houses subdirectories for the varies structure determination
and refinement tools and there associated data.
The TREE instruction provides a display of the tree structure.
The complete directory tree for a given compound compound can be
removed either from buttons on the SYSTEM-S menu or via the command line
instruction:
s compound remove
Example: s sdemo.ins
E.g. Shelx data (i.e. shelx.hkl and shelx.ins) for compound s1000
are stored in /mnt/shxdata/s1000
S will find the data (shelx.ins & shelx.hkl) when started up with s s1000
The default location can be changed by setting the environment variable SHXPATH
to the proper alternative.
System S is started in this context as s s1000
An alternative is to create an 's1000' sub-directory under 's' and copy
the CAD4 data as s1000.cad into '~s/s1000'.
System S is started as s s1000.
Example1:
sfun1.ins and sfun1.hkl
Example2:
sfun2.ins and sfun2.hkl
Example3:
sfun3.ins and sfun3.hkl
Example of an inorganic compound Cs2TiSi6O15:
csti.ins and csti.hkl
The transfer of the current structural results to the suggested space
group is effected by clicking on 'TRMX' and 'SPGR'.
The Formula and Z can be adapted when desired following this transfer.
Refinement can now proceed in the new spacegroup.
Example: Solve the 'sdemo' structure not in P21/c but in Pc (# 7).
At the anisotropic refinement stage, a message 'M/P P21/c' in RED
will appear to attract attention to the possibly missed or
pseudo-symmetry.
s demo.fcf
S will convert the data in the .fcf file into a shelx.hkl (HKLF 4
format) file and ask for additional missing data.
The .hkl file can be found in the subdirectory 'hklf'.
A shelx.ins file is prepared from the data available in the 'fcf'
file. Since an 'fcf' file doesn't contain information on the composition
composition, this info should be given manually.
Alternatively, when both a 'CIF' and an 'FCF' (either with extension
'.hkl' or '.fcf' is available, S can we invoked with:
s demo.cif
The KappaCCD/Denzo software provides two output file formats
SYSTEM-S can be started for this dataset via platon -s
import.cif.
Warning: Not all options available yet in this version !!!
The VIEW keyword invokes a display function giving a detailed
view on the reciprocal lattice (completeness etc.)
CRUNCH is in particular useful for light atom structures. It has solved
structures that turned out to be difficult for SHELXS and SIR.
DELABS is suggested by S in the isotropic refinement stage (and can be
ignored when desired by asking for 'SHELXL ISO' as the next calculation.
DELABS can be repeated in any subsequent refinement stage. It always
starts from the primary reflection file as supplied (with direction
cosines). Anisotropic thermal parameters are automatically converted
into corresponding isotropic U(eq) values before the DIFABS calculation.
It is a good idea to repeat the DELABS procedure when all scattering
power is accounted for (including H-atoms).
DIRDIF may have problems with structure determinations run with an
incorrect CONTENTS formula, in particular when the number of heavy atoms
is different from the number suggested.
Atom types are assigned to the resulting peaklist on the basis of
the contents formula.
The correct identification of a peak as C,O or N may be hampered by
difference in thermal parameters (i.e. periferal O atoms may fit the
peak height of a carbon atom and a central C may fit the peak height of
an O atom.
In case no a-priori information is available, C1H2 may be used as a
preliminary guess.
It provides information to be used for RELINK.
Current options are: ISO, ANISO, HATOMS, WEIGHTS and the number of
refinement cycles (default = 5 cycles).
Example: SHELXL ISO 3
Alternatively, SHELXL ISO 0 will provide a difference map that can be
used in the cycle of model completion as an alternative for EXOR and
EXORS.
The Default (Return) will suggest a suitable value (to be confirmed or
overruled) giving reasonable density and volume-per-atom values.
1 - WHAT IS 'SYSTEM-S'
(This should be changed in crunch.uni, line 490)
2 - IMPLEMENTATION REQUIREMENTS
It is assumed by default that all programs called from PLATON/S are
in the path so that PLATON/S can find them.
NOTE:Run s preferably under 'csh' or 'tcsh'. Problems have been
reported for REDHAT-6.0/bash. (REDHAT-5.2/bash o.k.)
3 - DEMO run(s) of S for C(10)H(12)O(6)
Assumptions:
sdemo.ins contains relevant information for a small organic compound
(with formula C10H12O6), on the cell, the wavelength
,info on the expected cell-content and HKLF 3 or 4 type, all in SHELX-style.
TITL SDEMO
CELL 0.71073 4.0007 7.7300 16.7597 90.0540 94.0760 90.0528
ZERR 2 0.0010 0.0008 0.0022 0.0096 0.0160 0.0144
SFAC C H O
UNIT 10 12 6
HKLF 4
S will return to the status where it was when restarted.
's sdemo'.
'rm -r sdemo'
4 - Worked Example in the Guided Mode
s[SHELXL ISO].
5 - DIRECTORY STRUCTURE of SYSTEM S
S operates in a directory structure under 'CURRENT_DIR/s/' (or '~USER/s/').
Note: '~USER/s/' is always used once this directory exists (either
deliberatery or as a result of a user error)
(Note: TREE does not display hidden files (stating with .) that
should not be touched, since they perform 'memory'-functions)
6 - PRIMARY (RAW) DATA
S provides three options for the storage of the original (raw)
diffractometer data from which S can be started.
7 - OTHER TRY-IT-YOURSELF EXAMPLES
Following are adapted (i.e. no prior space-group information given
ins the '.ins' data-file)
datasets
supplied with Dr A.D. Hunter's
SHELXTL course.
8 - How to Implement the new Space group suggested by ADDSYM
ADDSYM (either run explicitly or implicitly) will suggest an alternative
spacegroup in RED on the main S window. Addsym is invoked automatically
(unless switched off) at the start of the anisotropic refinement.
9 - How to run S on a 'CIF/FCF' Dataset
System S can be invoked using a 'fcf' (and 'cif') formatted file(s)
(e.g. taken from the IUCR-Acta Cryst C Web-page) via:
10 - KappaCCD/Denzo to PLATON/SYSTEM-S interface
11 - SUMMARY OF S-INSTRUCTIONS
ABSGAUSS
Absorption correction using a Gaussian Integration Grid.
ABSTOMPA
Absorption correction following the de Meulenaer & Tompa analytical
correction technique.
ASYM (VIEW)
Extensive listing and display of the averaging and completeness of
the reflection data as supplied.
AUTO
Synonymous for the No-Questions-Asked mode.
CRUNCH
A somewhat more time consuming Direct Methods alternative for SHELXS and
SIR.
DELABS
Empirical absorption correction following the modified DIFABS (Walker & Stuart)
approach.
DIRDIF
The method of choise for heavy atom structures.
EXOR
Work-up of the 'raw' peaklist of a structure determination package
(e.g. SHELXS).
EXORS
In function similar to EXOR but based on different techniques and
using parts of SIR for it.
FACE
Face as part of the description of the crystal for face-indexed
absorption correction.
FORMULA
Specification of the formula (e.g. C10H12O6).
HATOMS
Find H-atoms in difference map.
INVERT
Change absolute structure.
HELENA
Local data-reduction program for CAD4-data.
LIST
Refresh parameter status listing.
LOG
This option provides a log of the previous calculations done for this
compound.
MU
In of Mu value (mm-1) for absorption correction.
NQA
This is the No-Questions-Asked mode to operate S. This option may
be used to see whether a default structure determination leads to
interpretable results. If not, various other options should be tried
including solution in alternative spacegroups with alternative
structure solution techniques.
PLATON
Molecular geometry and other tools.
PLUTON
Molecular Graphics. Also used for the display of (intermediate) results,
atom renaming and the introduction of HFIX-ed atoms.
RELINK
Go to earlier context. See log-file.
SHELXL
Least-squares refinement on F^2.
SHELXS86
Alternative for the lastest SHELXS (SHELXS86 occasionally solves
structures that turn out to be more difficult with the latest version).
SHELXS
Default (Current version SHELXS-97-2) (I.e. fast) choice for the phase
determination of light atom structures
SIR
SIR97 provides an excellent alternative for SHELXS. It is slower but
often gives results with large poorly reflecting (low resolution) data
sets.
SPGR
By default, a list of spacegroups consistent with the current lattice,
Laue symmetry and systematic extinctions is presented with an indication
of a plausible first choice.
TRMX/TRNS
Transformation matrix (direct axes) for the transformation of the
supplied reflection data to the desired lattice. By default, a selection
may be done from a list of possibilities.
Z
Z times FORMULA should specify the unit cell content. Z is not
necessarily equal to the number of symmetry operations. It can be more
or less.
10-Jun-2002 A.L.Spek
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}