
![]()
![]()
Freely available software tools for chemical crystallography.
Lachlan M.D. Cranswick (L.Cranswick@dl.ac.uk)
CCP14 Project for Single Crystal and Powder Diffraction
http://www.ccp14.ac.uk ccp14@ccp14.ac.uk
|
|
Freely available software tools for chemical crystallography. Lachlan M.D. Cranswick (L.Cranswick@dl.ac.uk) CCP14 Project for Single Crystal and Powder Diffraction http://www.ccp14.ac.uk ccp14@ccp14.ac.uk |
|
Introduction
Crystal Structure Databases
Indexing/Spacegroup Assignment
Absorption Correction Options
Structure Solution
Structure Refinement
Single Crystal Suites
Graphically Interacting with the Structure
Fourier Map Generation and Viewing
Structure Quality Checking
Exporting/Importing CIF files
Specialised Programs
Structure Visualisation and Photorealistic Hardcopy Output
Solving Structures using Powder Diffraction
Utilities/Resources/PC Operating Systems
|
|
Freely available software tools for chemical crystallography. Notes on the Web Lachlan M.D. Cranswick (L.Cranswick@dl.ac.uk) CCP14 Project for Single Crystal and Powder Diffraction http://www.ccp14.ac.uk ccp14@ccp14.ac.uk |
|
One overhead every
45 seconds, 33 seconds, 25 seconds (new GNU Xtal Suite information from Thursday 29th Mar) or all is lost!Thus these notes with all the web links are on the web at:
http://www.ccp14.ac.uk/poster-talks/bca200/
Aim to try and give a feel for what is available but also what it is like to install and use many of the programs.
(by default, most of the windows programs install using a friendly setup.exe installer)
|
|
Freely available software tools for chemical crystallography. Disclaimer Lachlan M.D. Cranswick (L.Cranswick@dl.ac.uk) CCP14 Project for Single Crystal and Powder Diffraction http://www.ccp14.ac.uk ccp14@ccp14.ac.uk |
|
Note/Disclaimer:
If your favourite program or function has been missed, please feel free to mention this at the end of the talk. Subtle combination of time constraints and ignorance are probably the cause of this.Software that is freely available via the internet is emphasized even though others do exist and should also be investigated for those looking into building up their crystallographic toolkits:
NRCVAX
Like Xtal Suite – a Collaborative Community Effort.
(possibly available freely on the web soon(?))
E-mail Peter White (pwhite@pyrite.chem.unc.edu)
XSEED:
http://www.lbarbour.com/xseed
(US$ 1,000 for a site license)
B.A. Frenz SPD Single Crystal Suite
http://www.bafrenz.com
(US$ 995 for Academic Site License)
Hardware vendor based Single Crystal Software
|
|
Freely available software tools for chemical crystallography. Introduction Lachlan M.D. Cranswick (L.Cranswick@dl.ac.uk) CCP14 Project for Single Crystal and Powder Diffraction http://www.ccp14.ac.uk ccp14@ccp14.ac.uk |
|
If you are happy with the software you are presently using – why bother looking into alternatives?
Researchers are most at risk from not knowing what they do not know.
It is important to have access to a wide "genetic diversity" of software to handle wide variety of structure problems in the most effective manner. (for both the expert and part time crystallographer)
There is a "good" genetic diversity of freely available single crystal structure solution and refinement programs that can enabled both expert and "part time crystallographers" in tackling a wide diversity of structural problems.
i.e., struggling to solve heavy atom problems with direct methods programs where a program such as Dirdif can most likely solve and build the structure up to near completion at the click of a button.
Difficult equal atom problems can become solvable when using the available direct methods or fragment searching programs.
|
|
Freely available software tools for chemical crystallography. Introduction (cont’d) Lachlan M.D. Cranswick (L.Cranswick@dl.ac.uk) CCP14 Project for Single Crystal and Powder Diffraction http://www.ccp14.ac.uk ccp14@ccp14.ac.uk |
|
To enable fast download, where possible, software is mirrored at the CCP14 project web site via in the UK:
http://www.ccp14.ac.uk
and regional mirrors
(presently in Australia and Canada).
|
|
Freely available software tools for chemical crystallography. Newly Available Software Lachlan M.D. Cranswick (L.Cranswick@dl.ac.uk) CCP14 Project for Single Crystal and Powder Diffraction http://www.ccp14.ac.uk ccp14@ccp14.ac.uk |
|
Much newly released, freely available software have high quality "point and click" Graphical User Interfaces by default.
Freely available GUI Single Crystals SUITES are now becoming available and are extremely powerful in the manner that many crystallographic utilities are linked together with multiple pathways for solving and refining a structure. Thus no need for tedious manual file handling when passing between programs.
(ORTEX, WinGX, System S, GNU Xtal, CRYSTALS)
Crystal Structure Databases
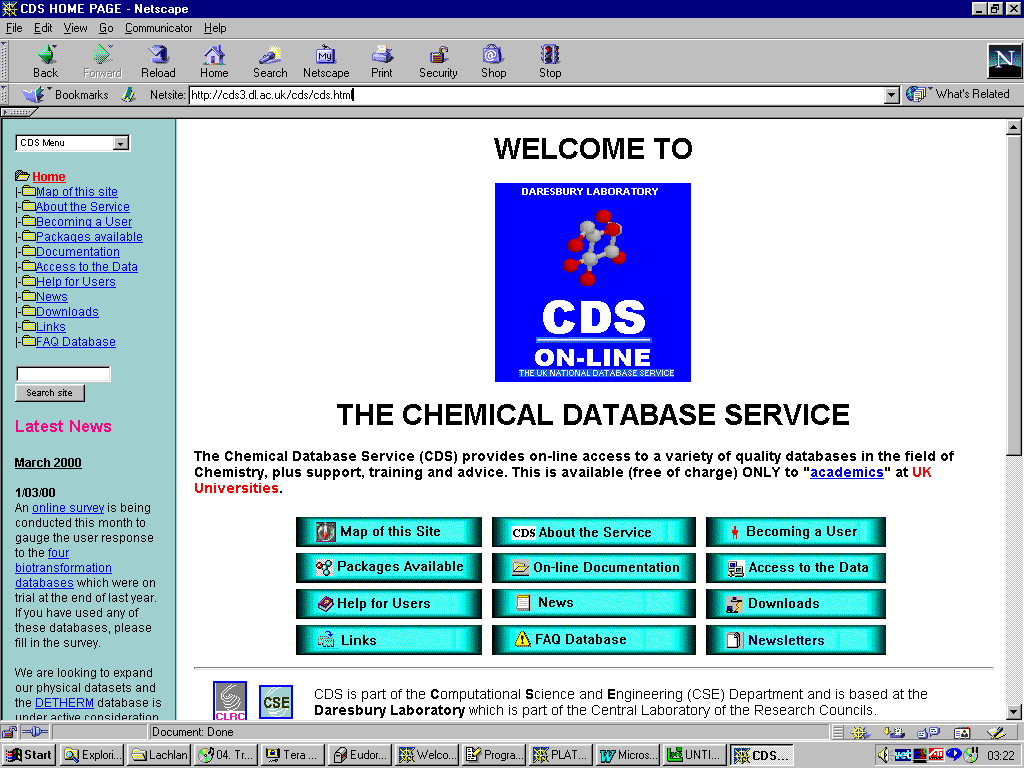
If you are a student or academic working in the UK, you have free access (via National Licences) to all the main Crystal Structure Databases via the EPSRC funded Chemical Database Service (CDS). You can register (no charge) at:
http://www.dl.ac.uk/CDS/cds.html
Single Crystal Indexing/Spacegroup Assignment
Most hardware vendors provide options for indexing but there are free-standing programs that can provide other options.
DIRAX for difficult Indexing problems.
ftp://ftp.chem.uu.nl/pub/dirax/
ABSEN Single Crystal Program by Patrick McArdle
(comes with the ORTEX and WinGX suites)
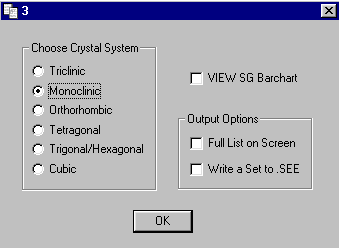
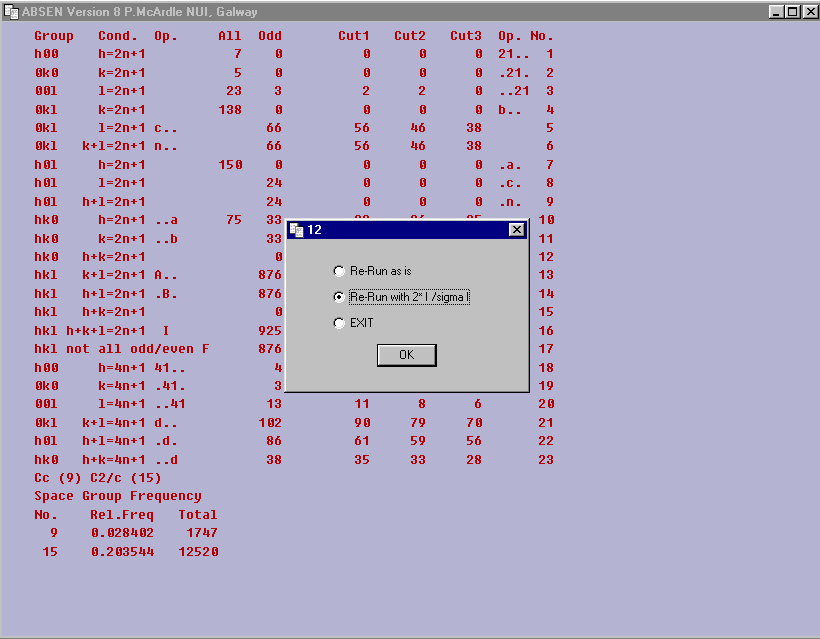
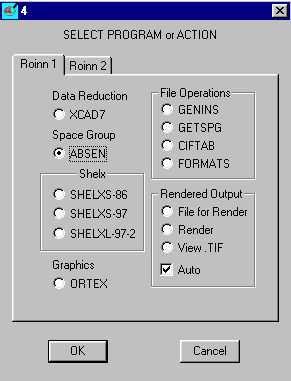
Indexing, spacegroup reasignment options within Platon/System S by Ton Spek for pre and after structure solution/refinement spacegroup assignment.
(Windows users: Platon is included with WinGX or can be run free standing)
Absorption Correction Options
Ortex, Crystals, WinGX and Platon give access to a variety of Absorption correction options.
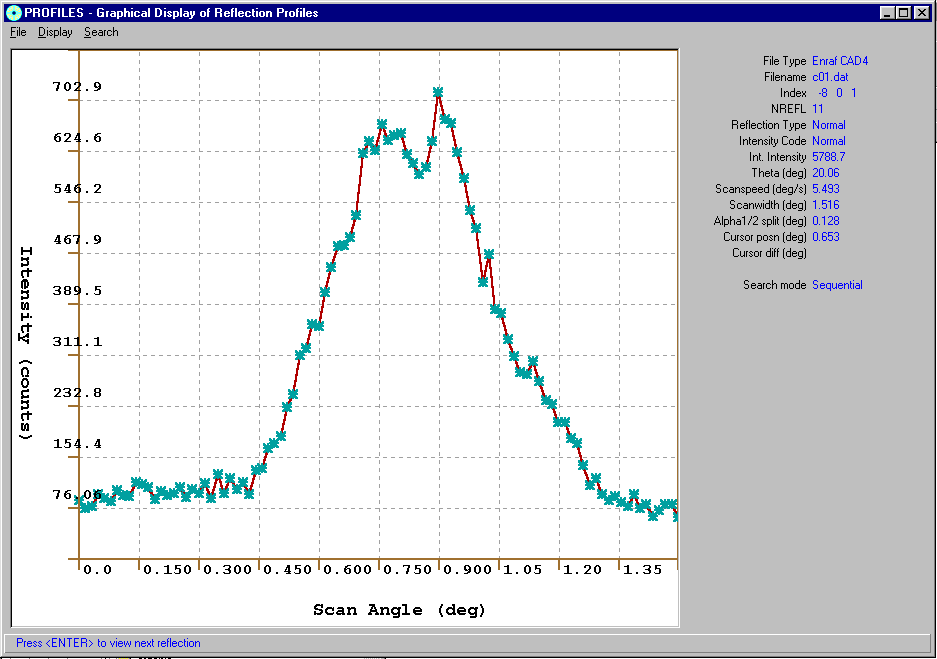
For example:
in WinGXViewing or HKL Profiles
Blessing DREAR Software
Numerical:
Gaussian, Analytical, Spherica l, Cylindrical
Semi-Empirical:
Psi-scans, Camel-Jockey, Multiscan
RefDelF:
Difabs, XABS2, Shelxa
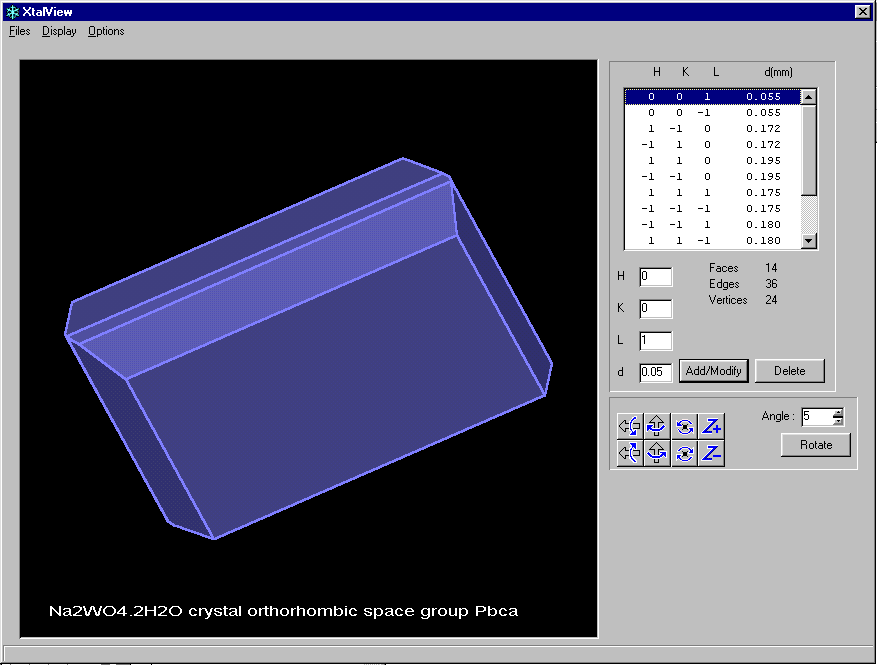
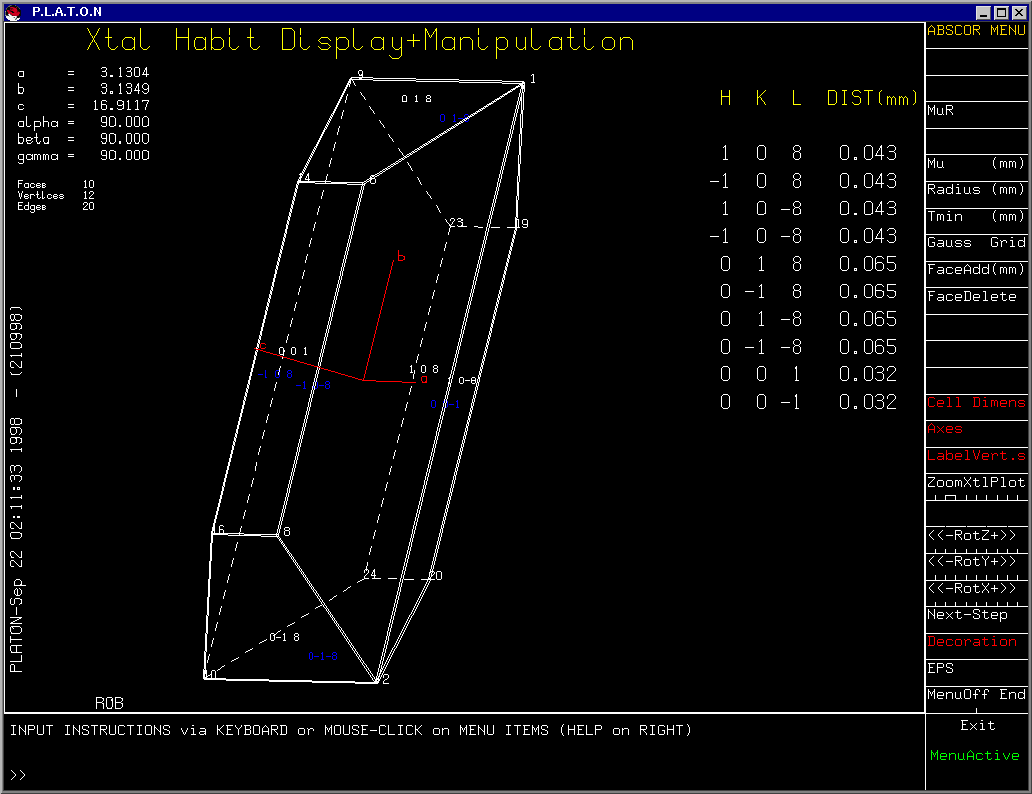
After defining faces, you can view the Crystal via
XtalView
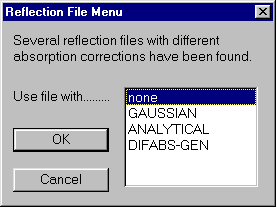
Before refinement, user is prompted which form of absorption corrected data to use:
Platon
(Has the option of Viewing the Crystal Habit)
Single Crystal Structure Solution
CAOS (also inside part of Sir97) – Ricardo Spagna, et. al.
Comes with an automatic Patterson Solution Option.
http://www.org.chemie.uni-frankfurt.de/egert/patsee.html
http://www.ccp14.ac.uk/ccp/web-mirrors/patsee/egert/patsee.html
Crisp – Part of the GNU Xtal Suite
Direct Methods
http://xtal.crystal.uwa.edu.au/
http://www.ccp14.ac.uk/ccp/web-mirrors/xtal/
Crunch – R. de Gelder, R.A.G. de Graaff & H. Schenk,
Direct Methods and automatic structure building
http://www-xtal.sci.kun.nl/xtal/documents/software/crunch.html
http://www.ccp14.ac.uk/ccp/web-mirrors/dirdif/xtal/documents/software/crunch.htmL
Dirdif - P.T. Beurskens, G. Beurskens, R. de Gelder, et al.
Patterson Methods for heavy atoms and fragments and automatic structure building
http://www-xtal.sci.kun.nl/xtal/documents/software/dirdif.html
http://www.ccp14.ac.uk/ccp/web-mirrors/dirdif/xtal/documents/software/dirdif.html
Windows:
http://www.chem.gla.ac.uk/~louis/dirdif/http://www.ccp14.ac.uk/ccp/web-mirrors/farrugia/~louis/dirdif/
Patsee – E. Egert and G. Sheldrick
Fragment Search
http://www.org.chemie.uni-frankfurt.de/egert/patsee.html
http://www.ccp14.ac.uk/ccp/web-mirrors/patsee/egert/patsee.html
Single Crystal Structure Solution (cont’d)
Shake’n’Bake (SnB) – Weeks, Miller, et al.
Dual-space direct methods. (Linux, SGI, Alpha executables via web)
http://www.hwi.buffalo.edu/SnB/
Shelxs 86/97- George Sheldrick
Direct Methods and Patterson Option
http://shelx.uni-ac.gwdg.de/SHELX/index.html
Sir92/97 – Sirware Group: Cascarano, Giacovazzo el al
Direct Methods and
automatic structure buildinghttp://www.ba.cnr.it/IRMEC/SirWare_main.html
http://www.ccp14.ac.uk/ccp/web-mirrors/sirware/IRMEC/SirWare_main.html
(Sir2000 is on the way – aim for handling difficult problems with more than 2000 atoms in the asymmetric unit)
Solver – in NRCVAX Suite – based on Multan
Direct Methods
Peter White (pwhite@pyrite.chem.unc.edu)
XFPA – Frantisek Pavelcik
Patterson Methods and automatic structure building
E-mail: pavelcik@fns.uniba.sk
Shelxs97 direct methods:
http://shelx.uni-ac.gwdg.de/SHELX/index.html
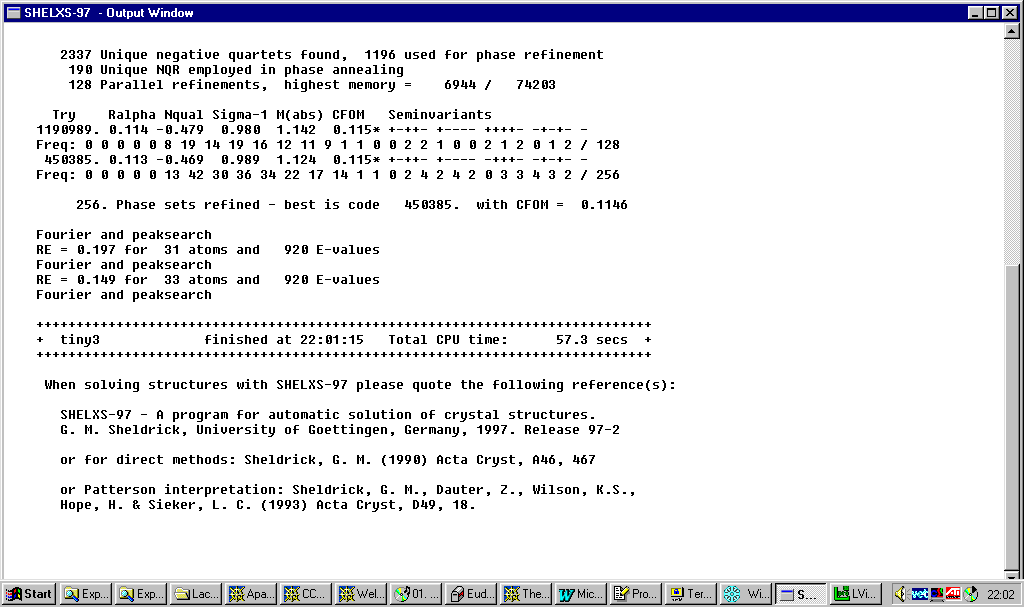
Resulting structure ready for building up:
Sir97 direct methods:
(Sir2000 is on the way – aim for handling difficult problems with more than 2000 atoms in the asymmetric unit)
http://www.ba.cnr.it/IRMEC/SirWare_main.html
http://www.ccp14.ac.uk/ccp/web-mirrors/sirware/IRMEC/SirWare_main.html
Here Sir97 is just at the point of automatically assigning atom types during the automatic building up of a structure:
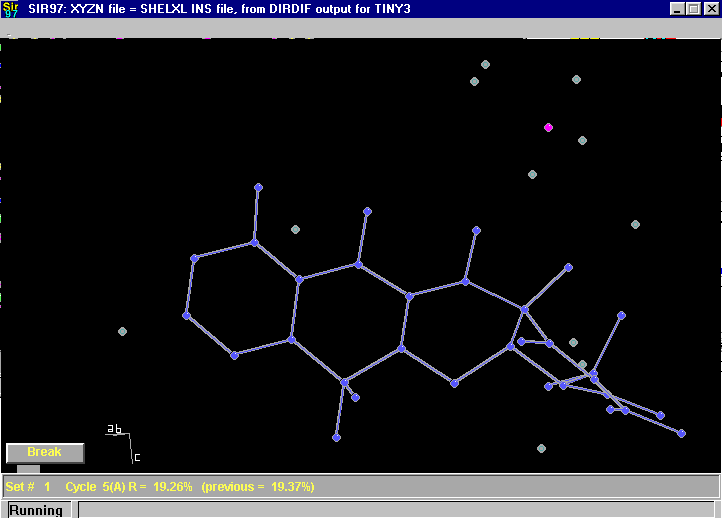
After Completion (just have to rename 3 atoms):
Dirdif
solving a Structure. Program of choice for heavy atom problems.http://www-xtal.sci.kun.nl/xtal/documents/software/dirdif.html
http://www.ccp14.ac.uk/ccp/web-mirrors/dirdif/xtal/documents/software/dirdif.html
Windows: http://www.chem.gla.ac.uk/~louis/dirdif/
http://www.ccp14.ac.uk/ccp/web-mirrors/farrugia/~louis/dirdif/
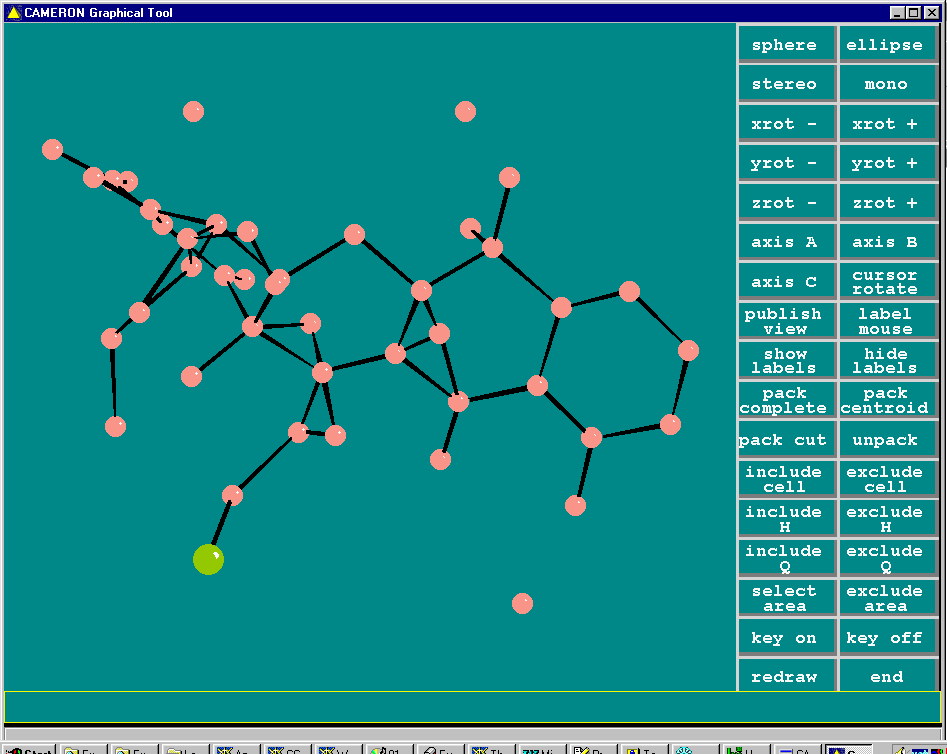
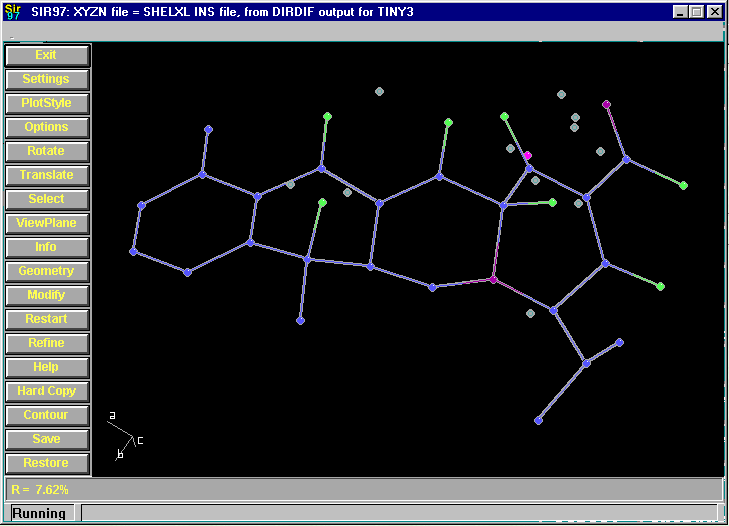
Resulting structure with every assigned non-Hydrogen atom correctly placed and named:
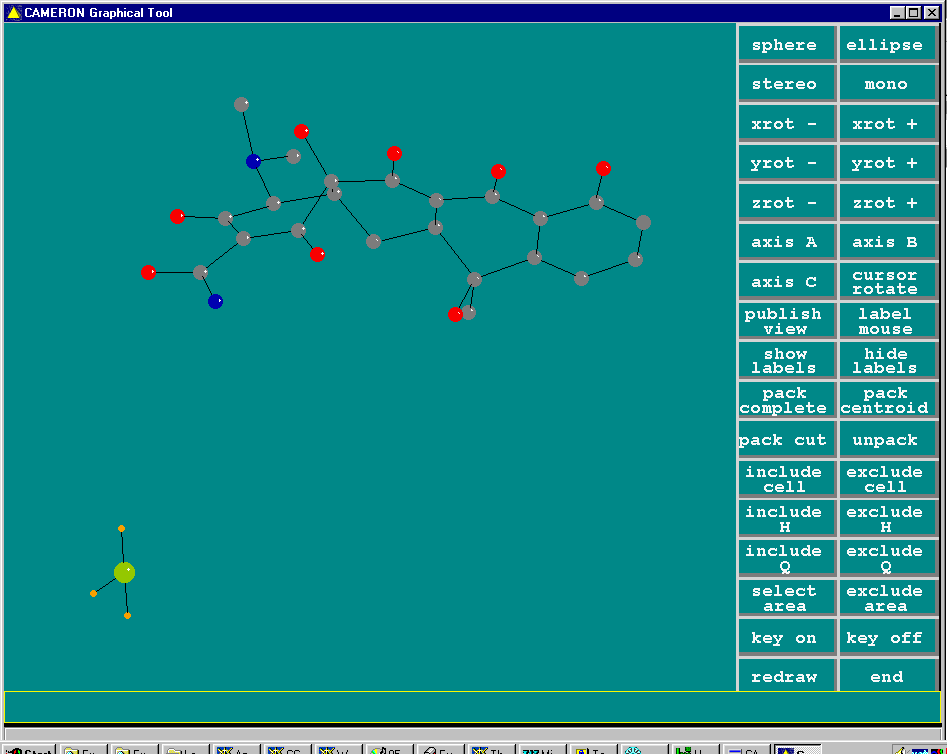
Crunch
Structure solved (via System S) and built to near completion (though this is really a heavy atom problem). One missing Carbon atom and various atom reassignments to be done.
(As shown via System S/Platon suite)
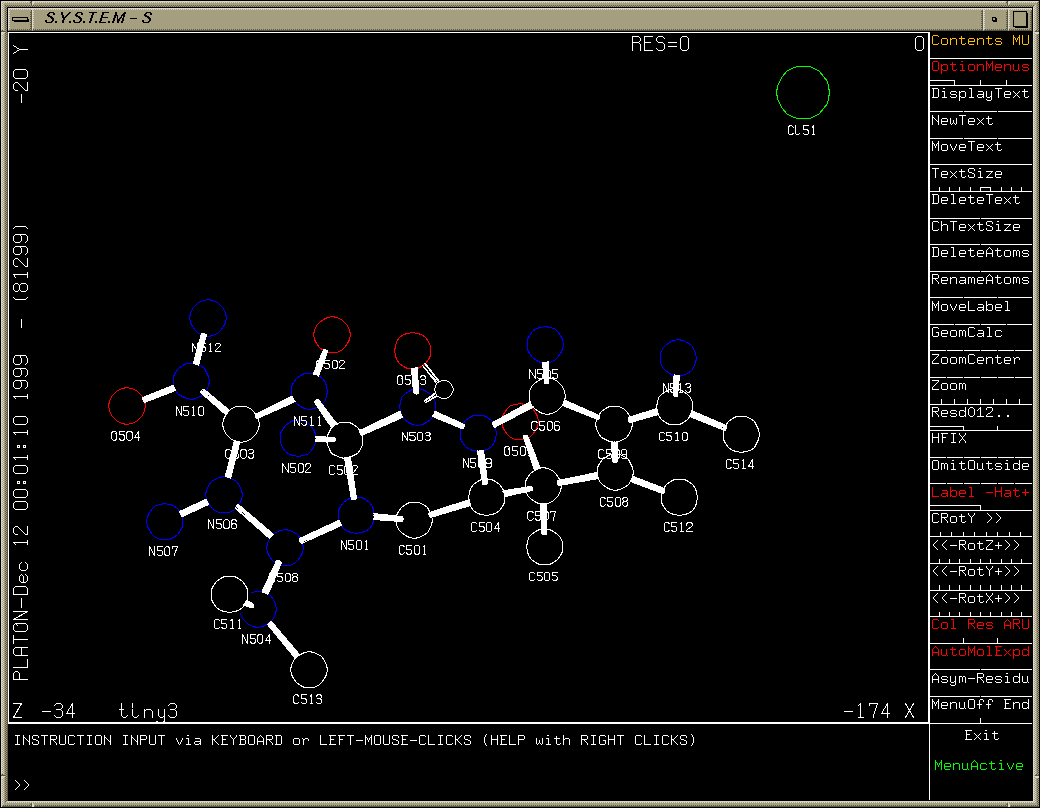
Special mention for
CRUNCH (UNIX):http://www-xtal.sci.kun.nl/xtal/documents/software/crunch.html
http://www.ccp14.ac.uk/ccp/web-mirrors/dirdif/xtal/documents/software/crunch.htmL
Reputation of being a Direct methods program of last resort. Succeeds where other direct methods fail.
Public example being the John Huffman "want to solve a structure" challenge on the sci.techniques.xtallography internet newsgroup (April 1994)CRUNCH does get some bad comments based on it reportely crashing UNIX workstations.
Personally have found CRUNCH to have a robust and problem free UNIX install/compile/usage.
Possible answer to above problems:
When crunch installer script asks for how much RAM you should give to Crunch,
can be a bad idea to state more physical RAM that you really have.
i.e., if you have 64 Meg RAM, state that Crunch can have 48 Meg.
Giving more RAM than physically available (with exception of FreeBSD and Digital UNIX) is great way to grind/crash the UNIX system to death. (near guaranteed to crash/freeze up SGI IRIX)
Tutorial Install: http://www.ccp14.ac.uk/tutorial/crunch/
"Trivial to use Crunch inside the System S suite."
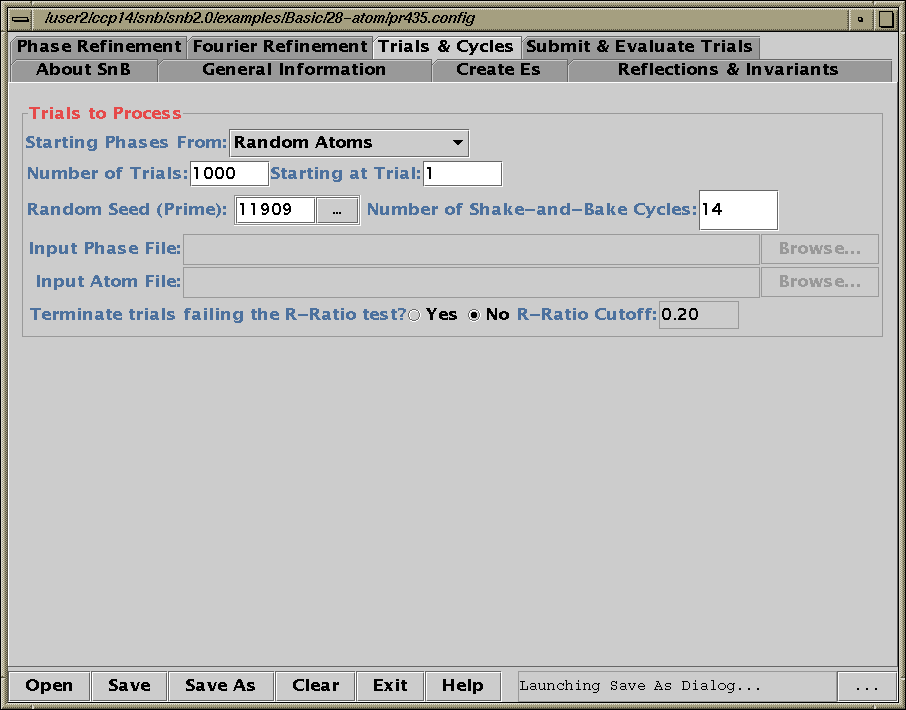
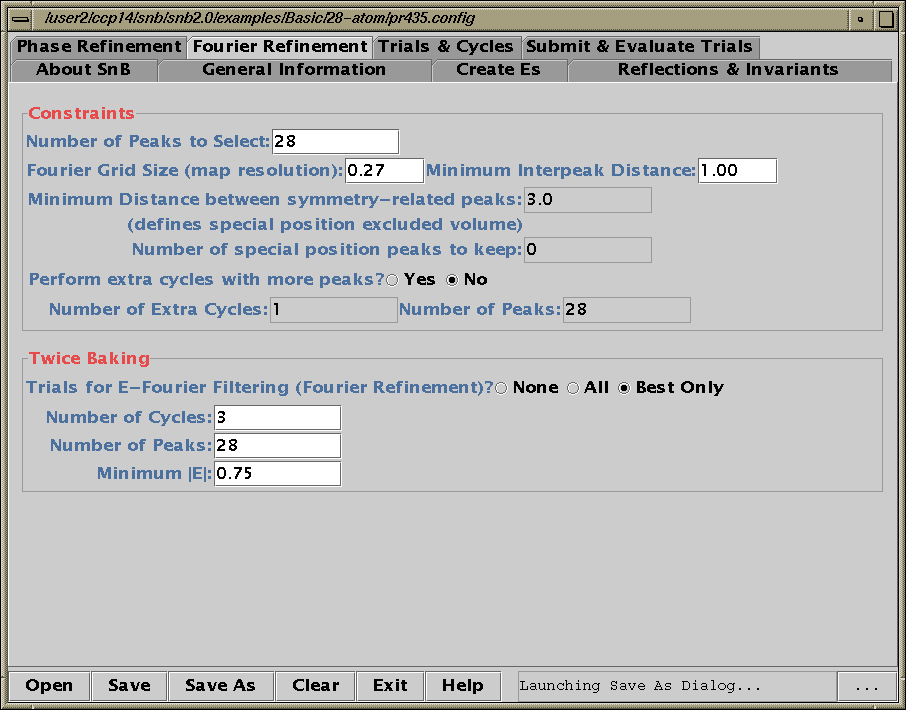
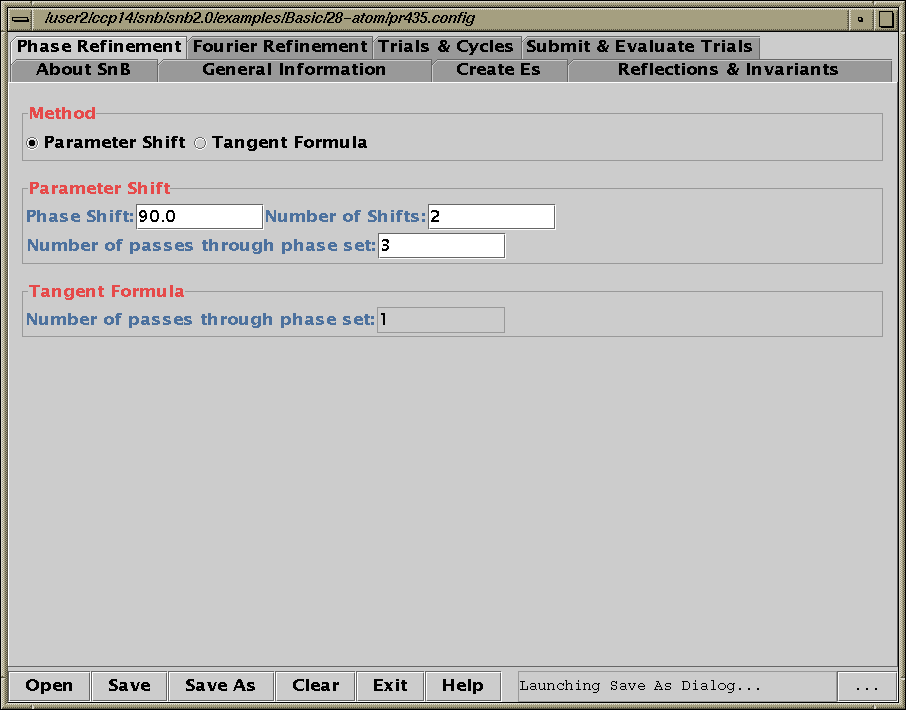
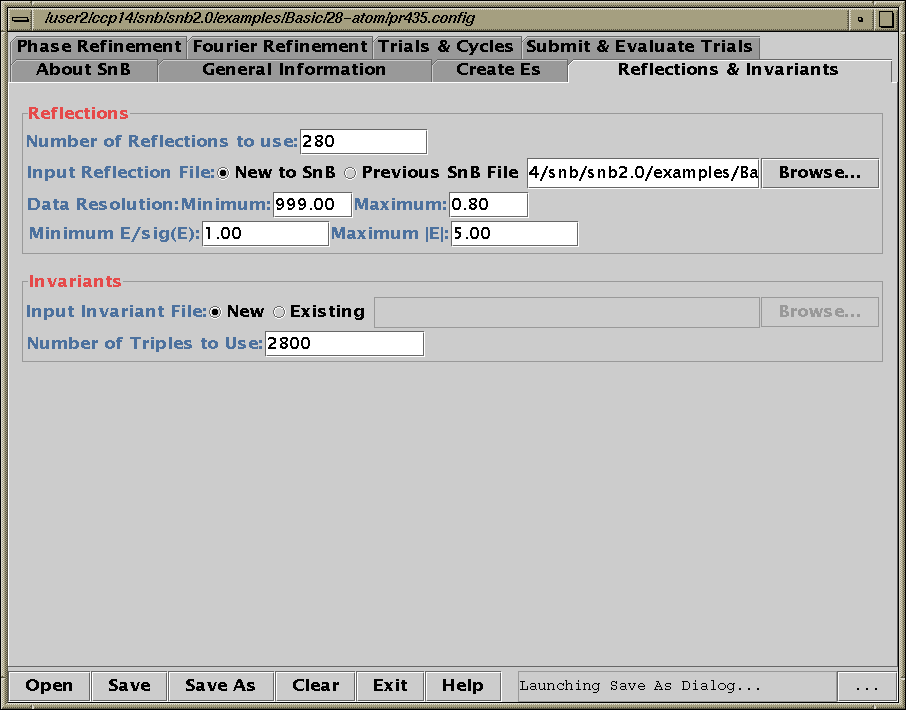
Shake’n’Bake (SnB) v2.0 Beta
(Linux, SGI and Alpha Executables available at present)
Download SnB via the web:
http://www.hwi.buffalo.edu/SnB/Availibility_snb_V2.htm
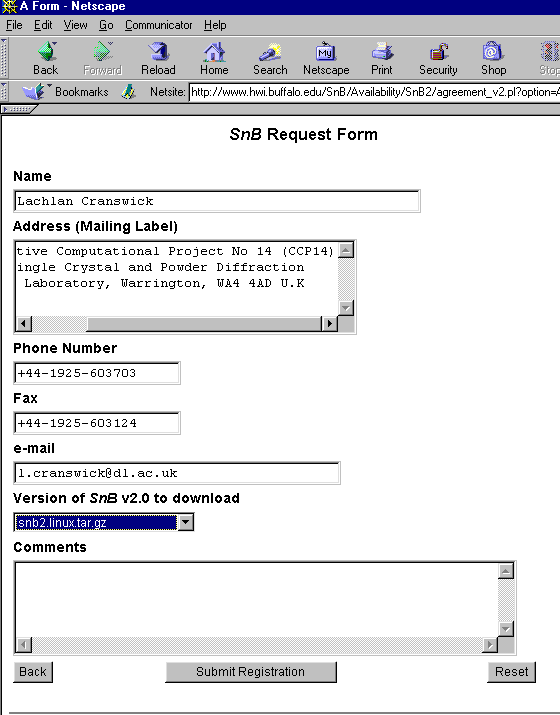 .
.
To extract the files, type gzip –d < filename.tar.gz | tar xvof –
Type Install to install (example from an SGI O2)
(Linux Java can be problematic – Dec 1999)
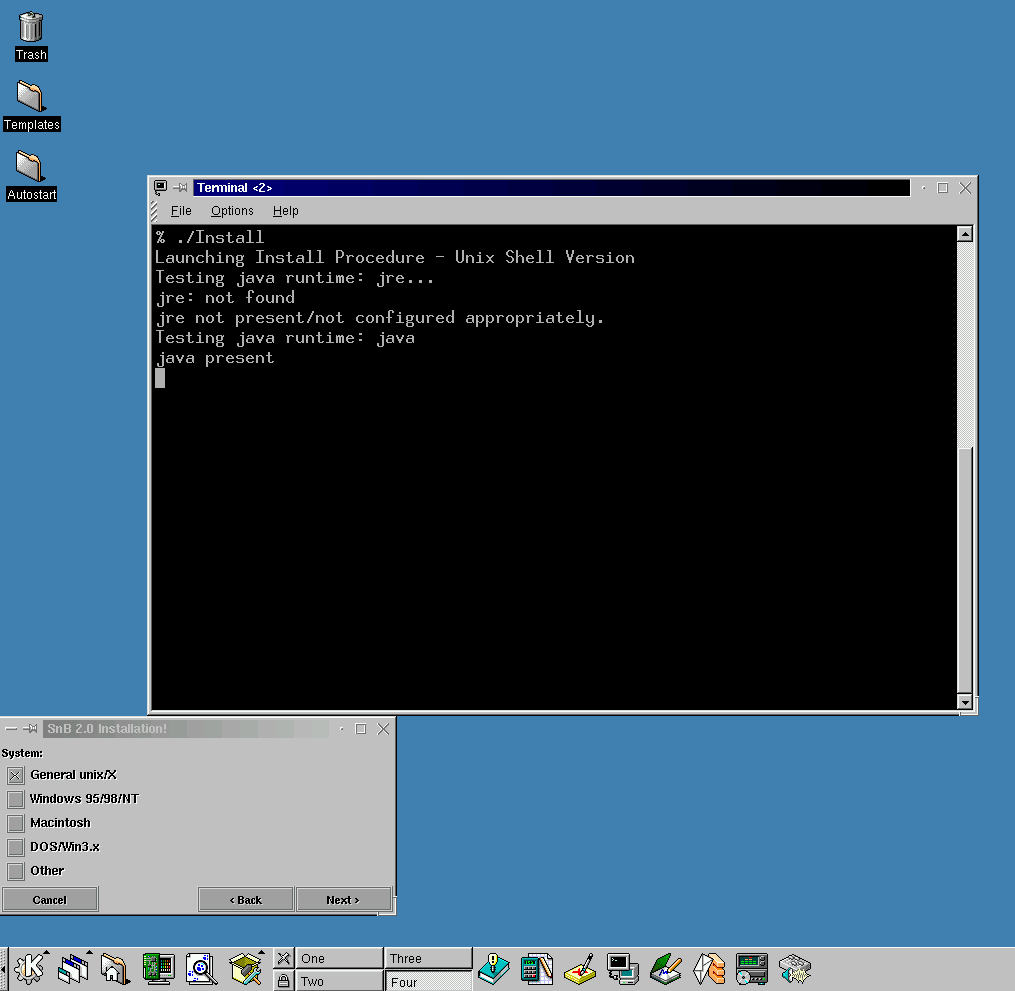
For Cshell, in the .cshrc
set path=(/snb_binary_directory $path)
do source .cshrc then rehash commands
And good times are now yours.
Running SnB (type SnB)
Java GUI Interface will then take you through setting up SnB up to visualising resulting structures. Though is not for those who like speedy results. Think "weeks/months" in computational time for large structures.
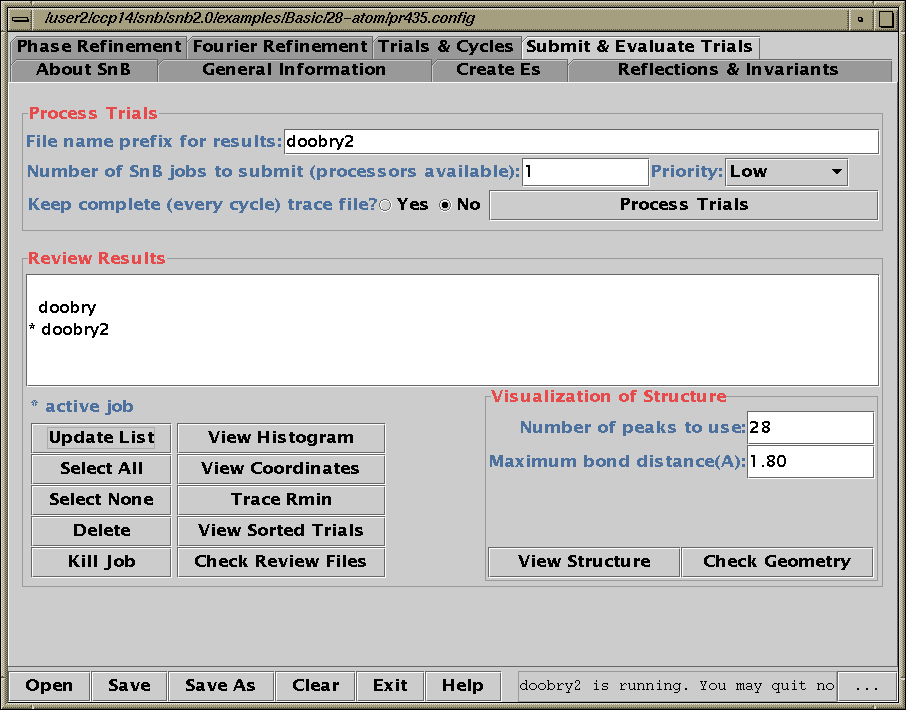
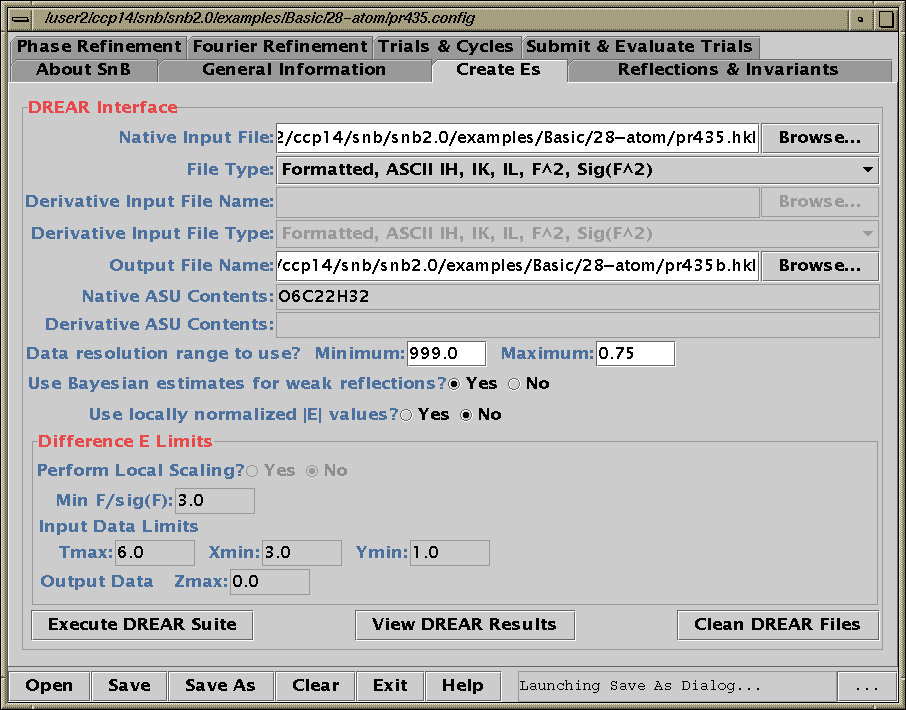
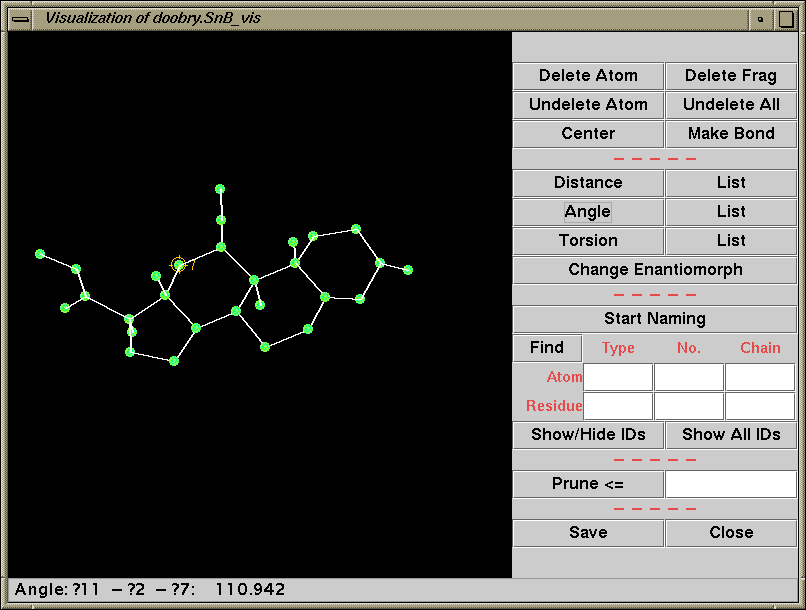
Difficult Structures/Collaborating with Software Authors
By default many difficult structures may not solve and/or show up problems with existing software. Obvious solution is to pass this on to structure solution software authors to help "train up" their programs:
(Not sure how many people are into this habit?)
Recent March 2000 Example from NRC Labs in Ottawa:
Cyclodextrin with guest molecules.
~300,000 unmerged reflections, ~80,000 merged.
Cell ~30 x ~30 x ~30. > 1000 atoms in the asymmetric unit
"Nothing solves it"
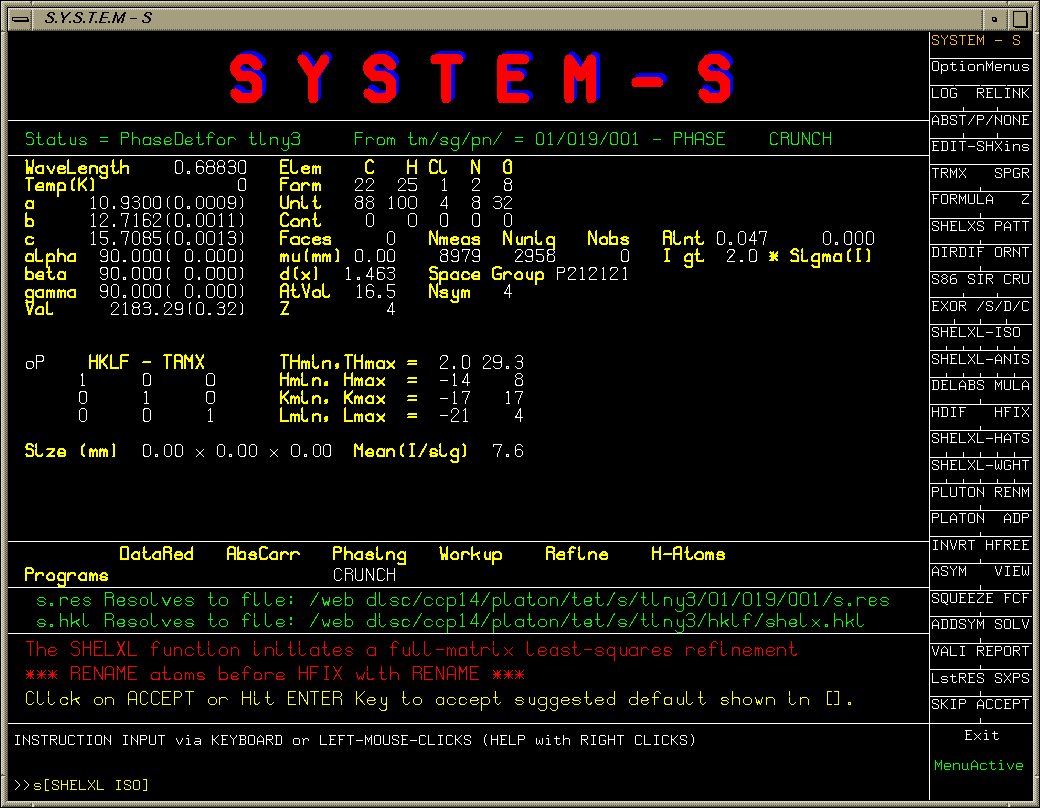
Installing System S with Crunch and Sir97 on an SGI O2 shows up problems/bugs with both direct methods programs. (Reported to authors)
Shake n Bake on SGI O2 = into its 40th trial after 2 weeks. Last heard: was still on the case (4 weeks later).
Within 2 weeks: Sir fixed and structure solved with Sir2000 in default mode. (expected release of Sir2000 at ECM 2000 meeting in Nancy)
New version of Crunch is to be available soon.
Request for Collaboration by the Sirware Group
29th March 2000: Invitation from the Sirware Group to collaborate on solving unsolved data/difficult structures you may have accumulated (up to and greater than 2000 atoms in the asymmetric unit)
(consider sending in your favourite solved and unsolved structures)
Carmelo Giacavazzo
(E-mail : c.giacovazzo@area.ba.cnr.it)
http://www.ba.cnr.it/IRMEC/SirWare_main.html
http://www.ccp14.ac.uk/ccp/web-mirrors/sirware/IRMEC/SirWare_main.html
Present range of solved structures with the alpha-test version of Sir2000 range from:
"202 non-H atoms" to
"1,910 non-H atoms (with 374 waters)"
2D-3D Model Building Software
(Applicable for Generating 3D fragments for Patsee/Dirdif Orient)
For a list, refer:
http://www.ccp14.ac.uk/solution/2d_3d_model_builders/
E.g., CORINA (COoRdINAtes):
http://www2.ccc.uni-erlangen.de/software/corina/free_struct.html
Comes with a Java Molecule Editor for building up the 2D structure over the web:
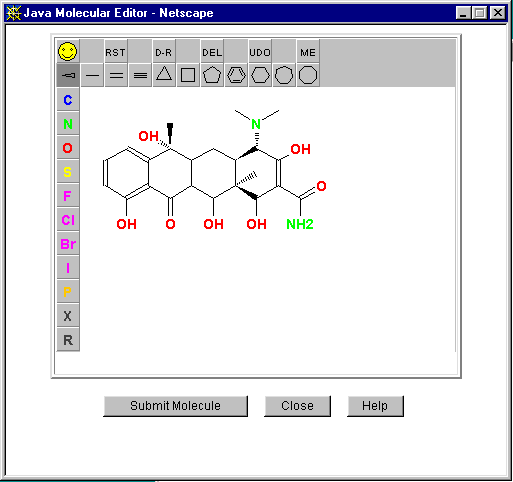
(Recommend using the "Direct Submission" option for immediate resulting MOL file, not the batch submission)
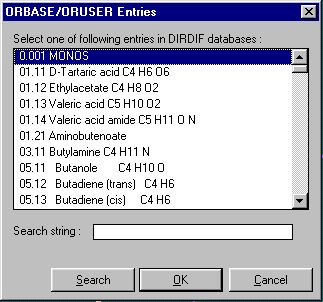
Getting Fragments into Dirdif for Windows.
New WinGX "SXGRAPH" GUI Shelx INS/RES file Editor
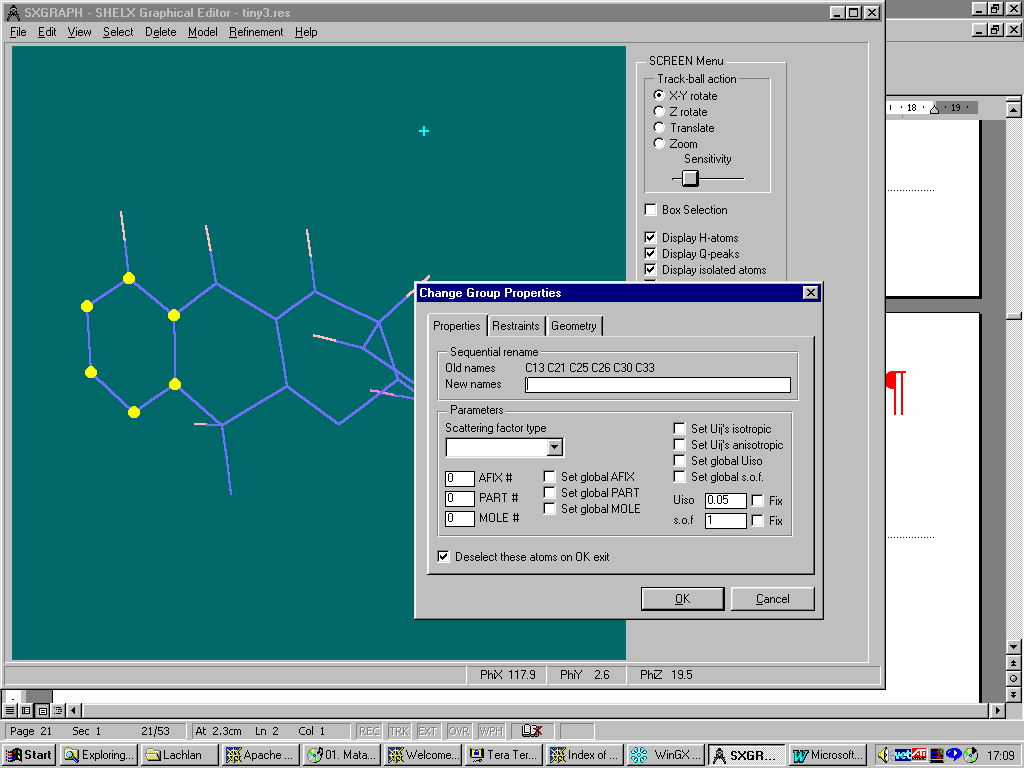
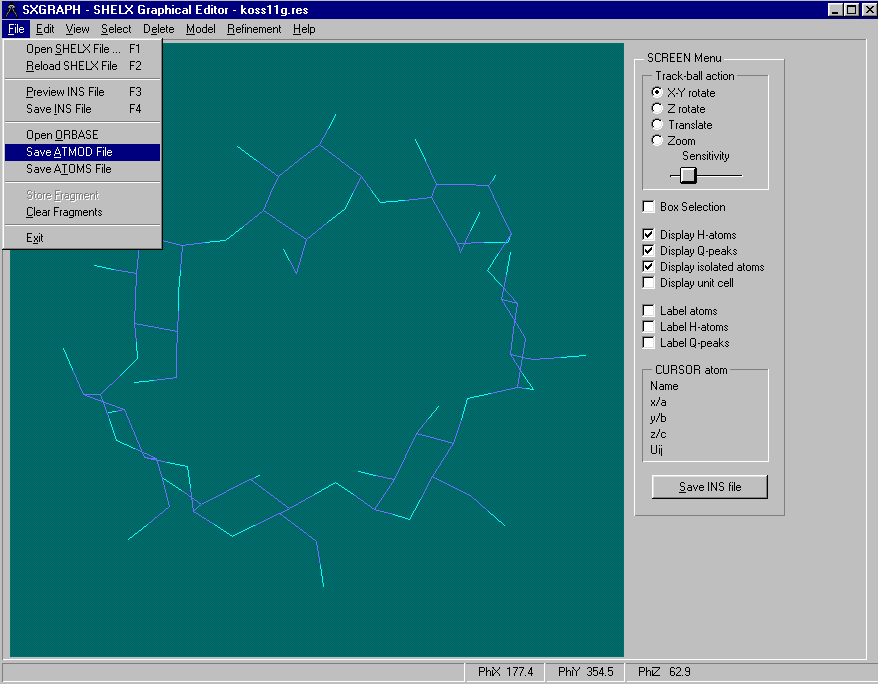
Either graphically Browse and Edit the Orbase Entries or Open an imported structure file (CSSR, CSD, Shelx or CIF from existing structure refinement),
clean it up, then save it as a fragment ready for immediate use with Dirdif for Windows. (or any Dirdif)(In the following case, a Cyclodextrin Fragment)
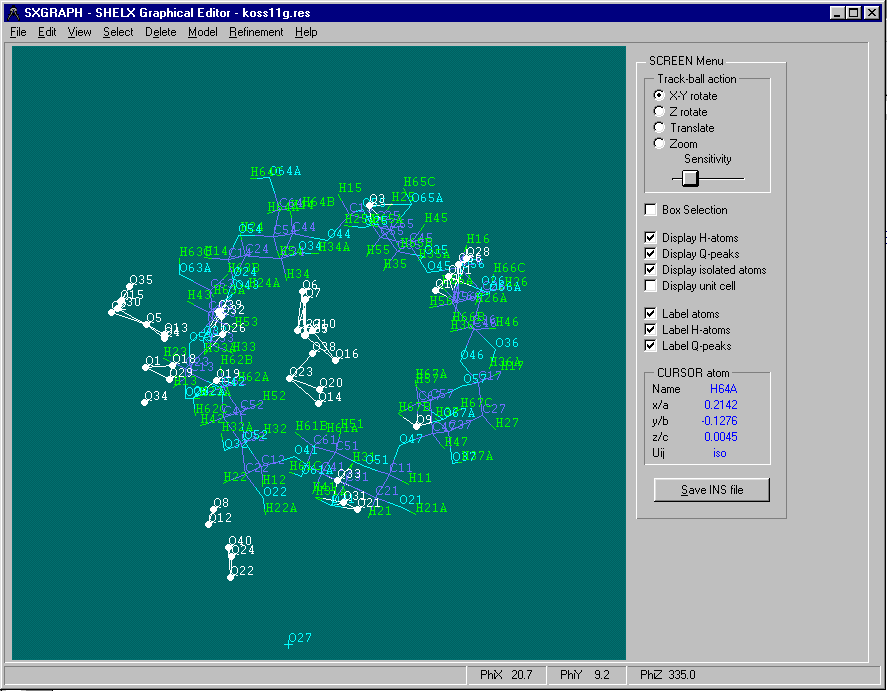
Single Crystal Structure Refinement
Shelxl (refinement program for System S/ORTEX/WinGX)
http://shelx.uni-ac.gwdg.de/SHELX/index.html
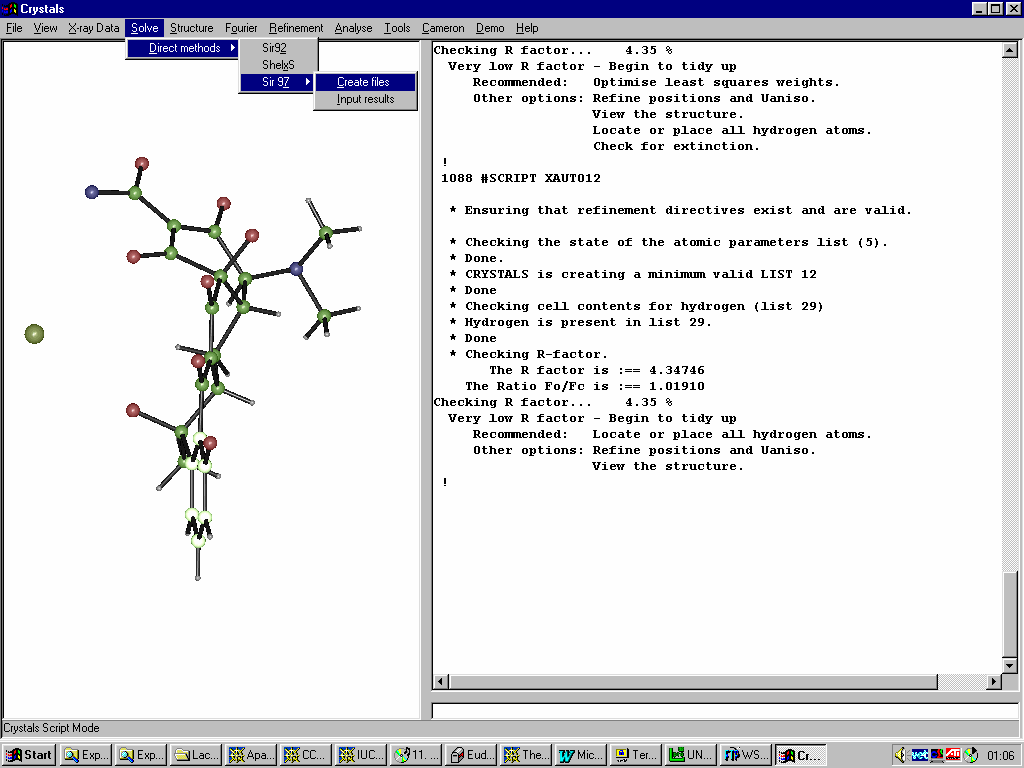
Crystals
http://www.xtl.ox.ac.uk/
http://www.ccp14.ac.uk/ccp/ccp14/ftp-mirror/crystals/pub/crystals/
CAOS (freestanding and also available inside Sir97)
http://www.isc.mlib.cnr.it/caos/
http://www.ccp14.ac.uk/ccp/web-mirrors/caos/caos/
http://www.ba.cnr.it/IRMEC/SirWare_main.html
http://www.ccp14.ac.uk/ccp/web-mirrors/sirware/IRMEC/SirWare_main.html
GNU Xtal
http://xtal.crystal.uwa.edu.au/
http://www.ccp14.ac.uk/ccp/web-mirrors/xtal/
(NRCVAX – Contact: pwhite@pyrite.chem.unc.edu)
Some more specialised examples:
JANA
http://www-xray.fzu.cz/jana/jana.html
http://www.ccp14.ac.uk/ccp/web-mirrors/jana/jana/jana.html
Incommensurate and f’ f’’ refinement
GSAS
ftp://ftp.lanl.gov/public/gsas/
http://www.ccp14.ac.uk/ccp/ccp14/ftp-mirror/gsas/public/gsas/
Combined powder/single crystal. f’ f’’ refinement.
Fullprof (is included with Winplotr graphics program)
ftp://bali.saclay.cea.fr/pub/divers/winplotr/
http://www.ccp14.ac.uk/ccp/ccp14/ftp-mirror/fullprof/pub/divers/winplotr/
Combined powder/single crystal. Incommensurate structures.
Example of Sir97/CAOS:
By default, CAOS is used via a powerful scripting language. It is more Photogenic when operated inside Sir97 (point and click mode) thus the following screen dumps are from Sir97
Deleting, re-naming, re-sorting, etc at the click of a button.
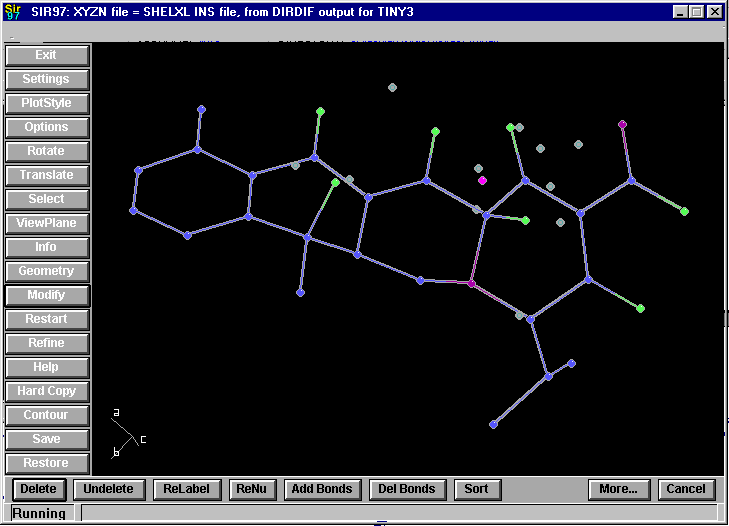
Automatic Addition of Carbon bonded hydrogens at the click of a button:
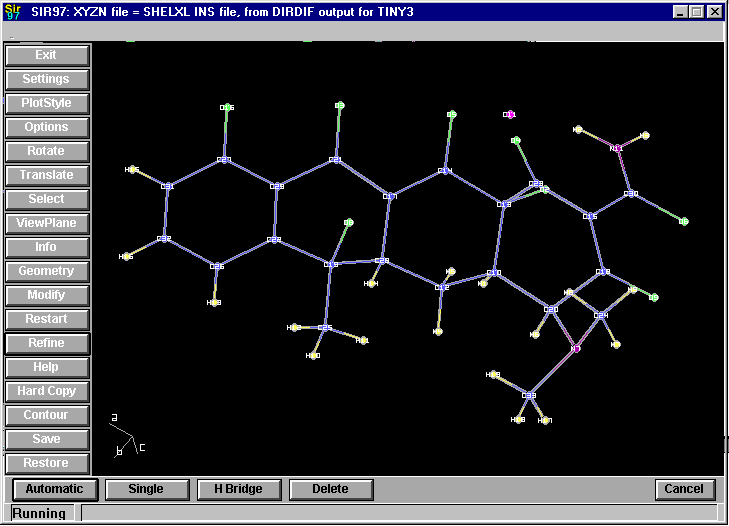
GUI CRYSTALS for Windows:
Guided refinement of a crystal structure including automatic hydrogen addition.
Overlay of calculated and found hydrogens for high quality hydrogen placement:
Restraints and Constraints defined by selecting atoms using point and click of the mouse:
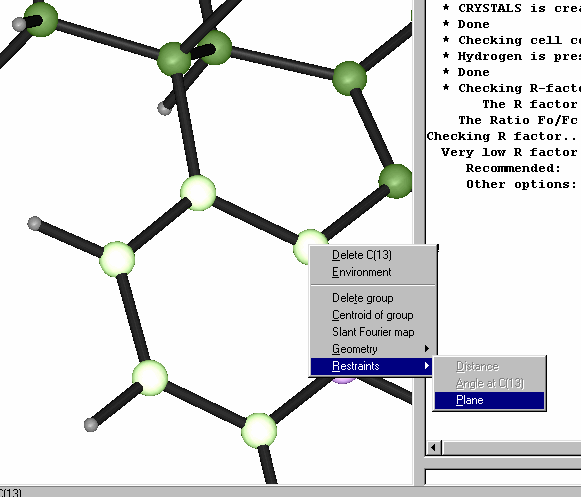
Single Crystal Suites
Single crystal suites can be extremely powerful as they allow users to quickly pick and mix a variety of programs at the click of a button.
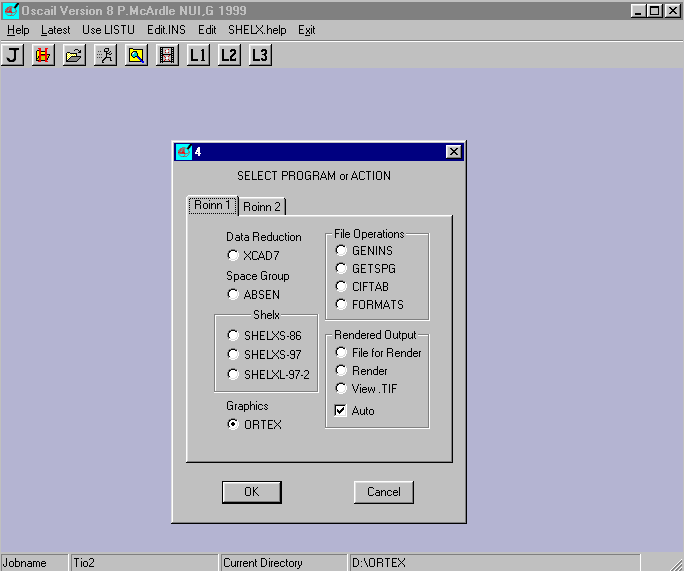
ORTEX - Patrick McArdle
Shelx86/Shelxs97 for Structure Solution
Shelxl for refinement
http://www.nuigalway.ie/cryst/
http://www.ccp14.ac.uk/ccp/web-mirrors/ortex/cryst/
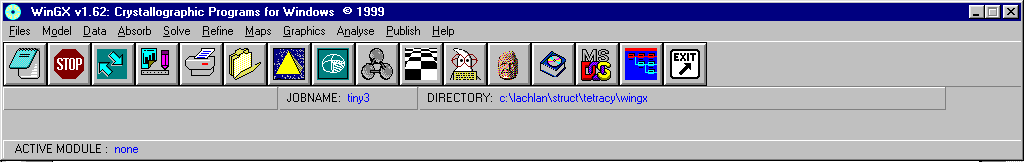
WinGX - Louis Farrugia
Shelxl for refinement
http://www.chem.gla.ac.uk/~louis/software/
http://www.ccp14.ac.uk/ccp/web-mirrors/farrugia/~louis/software/
System S/Platon - Ton Spek
Shelx86/Shelx97, Sir97, Dirdif, Crunch Structure Solution
Shelxl for refinement
http://www.nuigalway.ie/cryst/
http://www.ccp14.ac.uk/ccp/web-mirrors/ortex/cryst/
Crystals – David Watkin, Prout, Carruthers, Betteridge, Richard Cooper
Shelxs97 and Sir92/Sir97 for Structure Solution
Crystals for refinement
http://www.cryst.chem.uu.nl/platon/
http://www.ccp14.ac.uk/ccp/web-mirrors/platon-spek/platon/
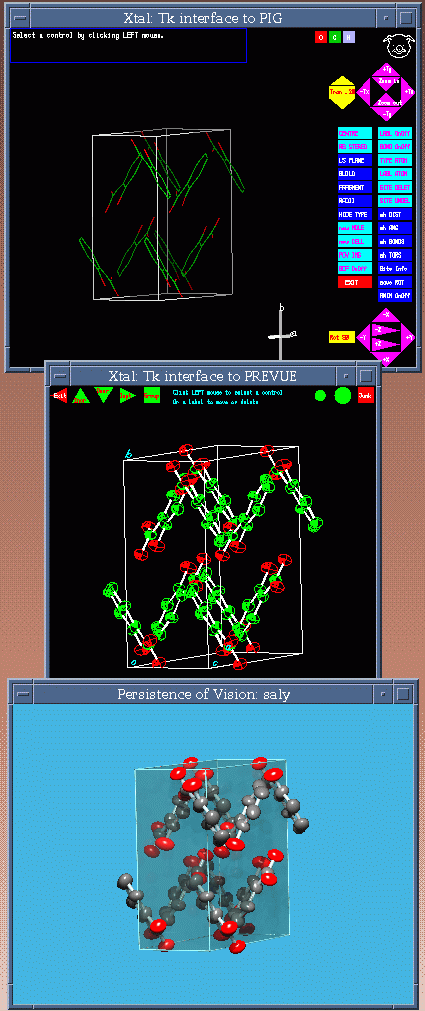
GNU Xtal –
Sid Hall, Doug Boulay, R. Olthof-Hazekamp and co-workers.Crisp, Patsee for Structure Solution
3 Separate Least Squares Refinement Programs within GNU Xtal
http://xtal.crystal.uwa.edu.au/
http://www.ccp14.ac.uk/ccp/web-mirrors/xtal/
NRCVAX – Peter White (E-mail: pwhite@pyrite.chem.unc.edu)
Solver for Structure Solution – others link in. NRCVAX for Refinement,
Properties of the Single Crystal Suites
Note: It is almost impossible to do justice to any of these single crystal suites withing the time allowed.
A characteristic is that they
very high quality Graphical User Interfaces (GUIs) and linking to wide variety of programs.You can be guided through the structure solution and refinement.
But you are not straight jacketted and can define your own route through the structure solution and refinement. It is entirely up to your personal preferrences and likes. And you can go between the suites if you wish
E.g.,
Face Index Absorption Correction using WinGX/Platon
Spacegroup analysis via ORTEX
Solving via WinGX
Building via System S/Platon
Refinement and Hydrogen placement in Crystals or GNU Xtal
Extra Structure validation and via WinGX, System S/Platon, GNU Xtal and Crystals
Photorealistic Rendering of any cavities in GNU Xtal
Rendering photorealistic images via ORTEX
Makes cross validation of results performed in different programs quite trivial.
Can save hours in manual file handling in going between different programs.
WinGX/ORTEX/System S/Shelx/CAOS/GNU Xtal/Crystals - all have their own nuances and strengths for interacting with the structure and data. This variety is not a bad thing as you can gain insights into the structure nuances/data quality via these differing routes.
Single Crystal Suites Interfaces
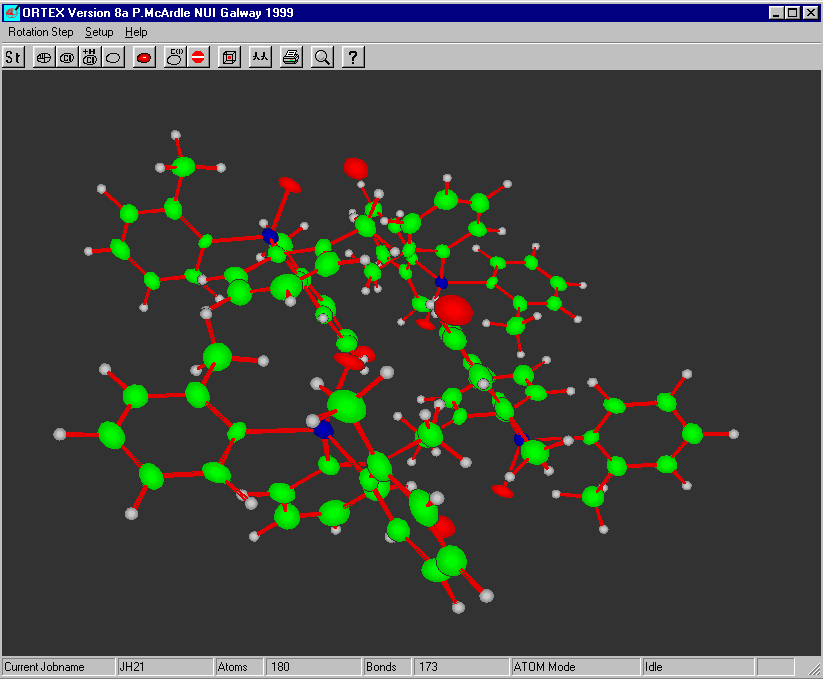
ORTEX:
WinGX:
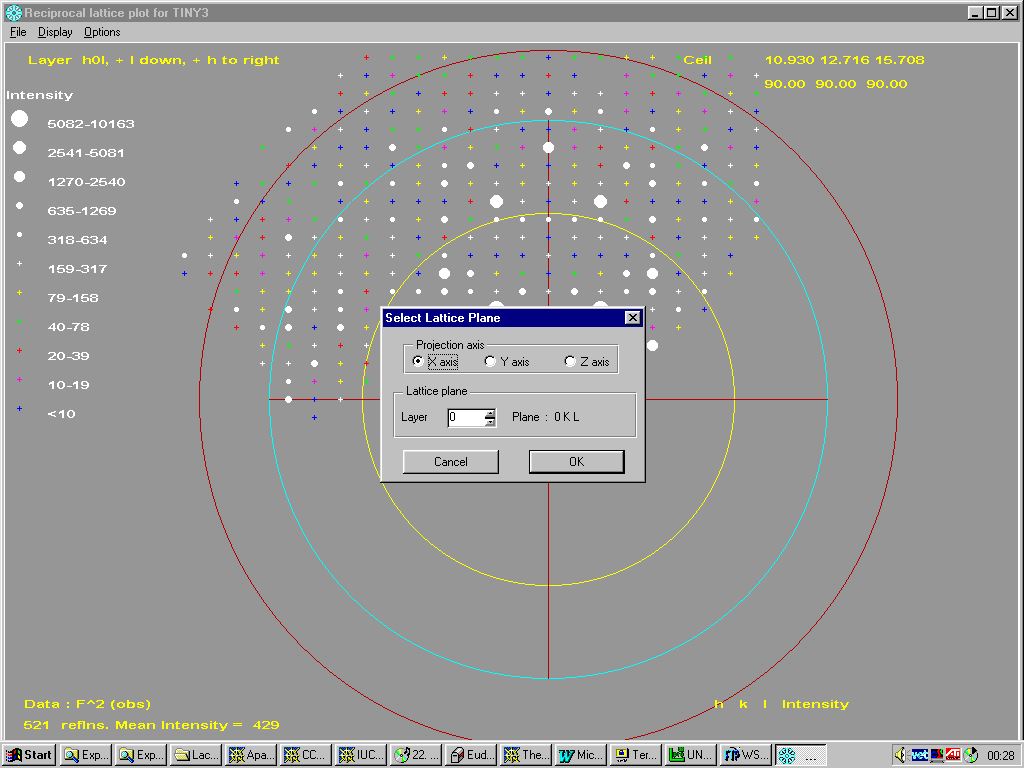
Platon/System S:
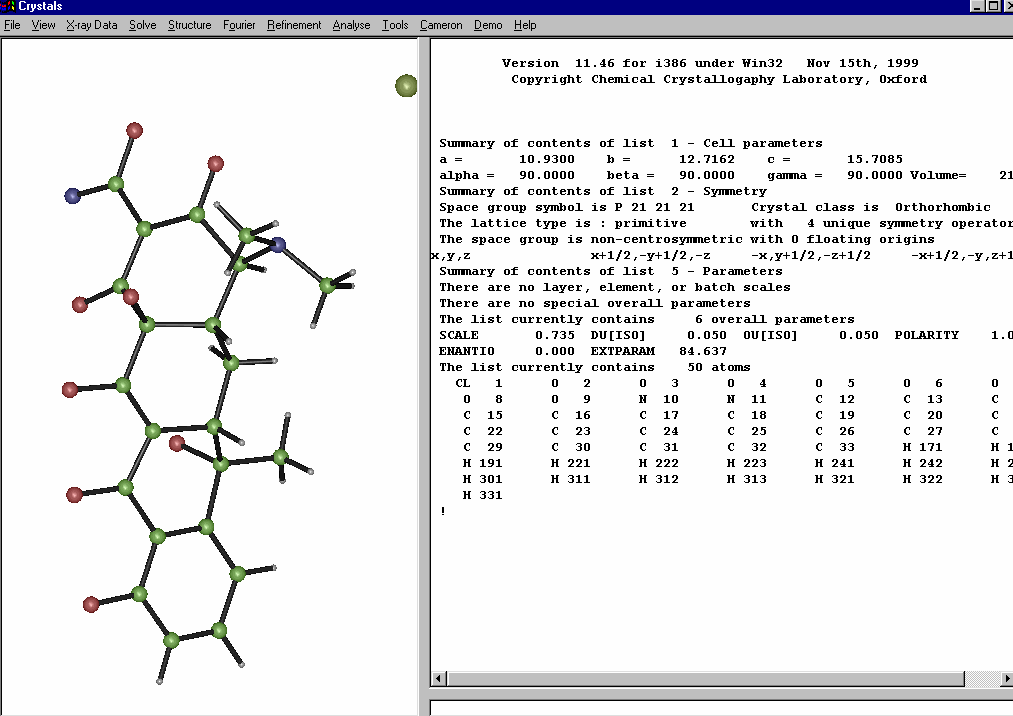
Crystals:
Example of the GNU Xtal Suite
Using the PIG (Portable Interactive Graphics) Interface
Non-Trivial Structure Solution of an Inorganic made Trivial using WinGX
(A Cesium Titanium Silicate –
relevant to nuclear waste storage)(Note: there is a difference between a program’s ability to solve a structure and a person’s ability to use a structure solution program)
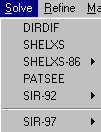
WinGX gives access to 6 structure solution programs at the click of a mouse button.
Try solving using
default Shelxs97 – no recognisable solutionTry solving
default Sir97 – no recognisable solutionTry solving using
default Dirdif –Structure solved and auto-builds to completion
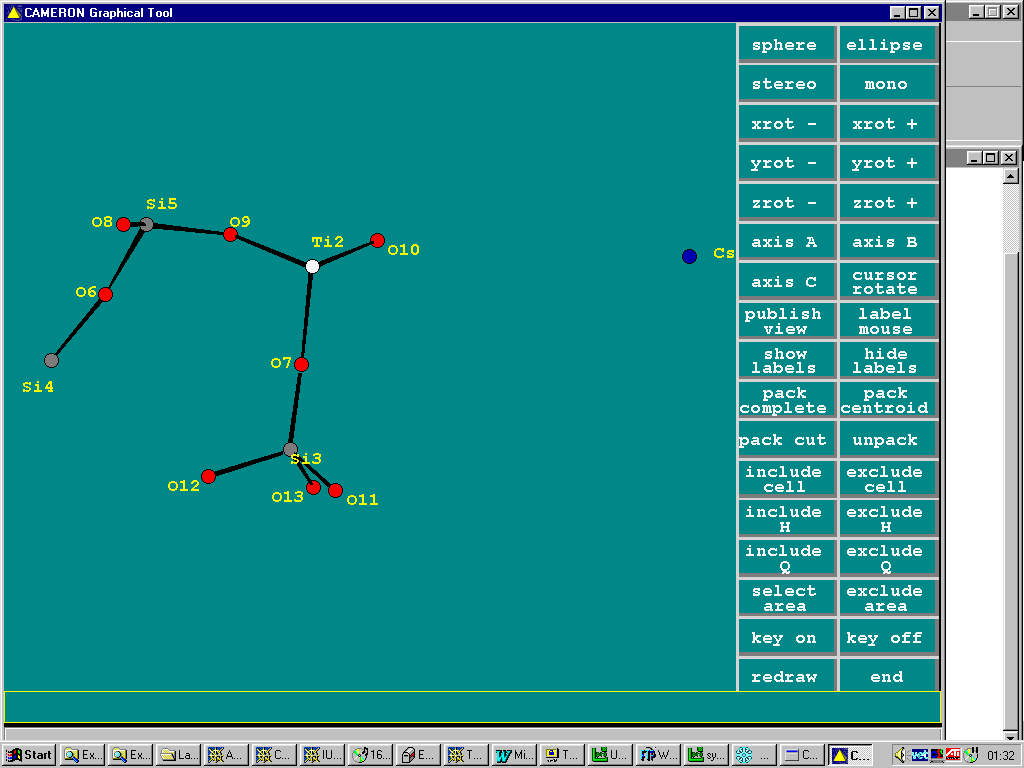
Multiple Pathways for Structure Solving
Solving in one program – automatic building in another – via System S
(Again, Cesium Titanium Silicate)
Solve in Shelx86
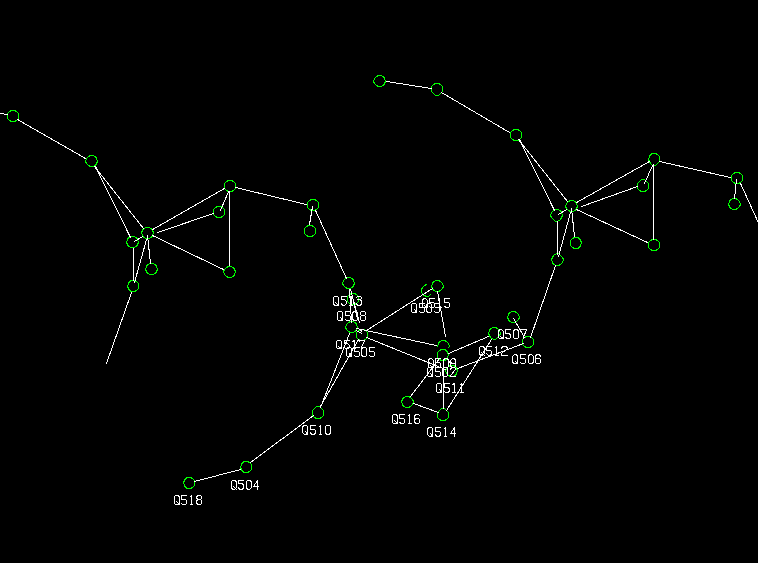
Use the System S/Platon EXOR function to build up the structure
(also have the other options of using Crunch, Sir and Dirdif to build up the structure).
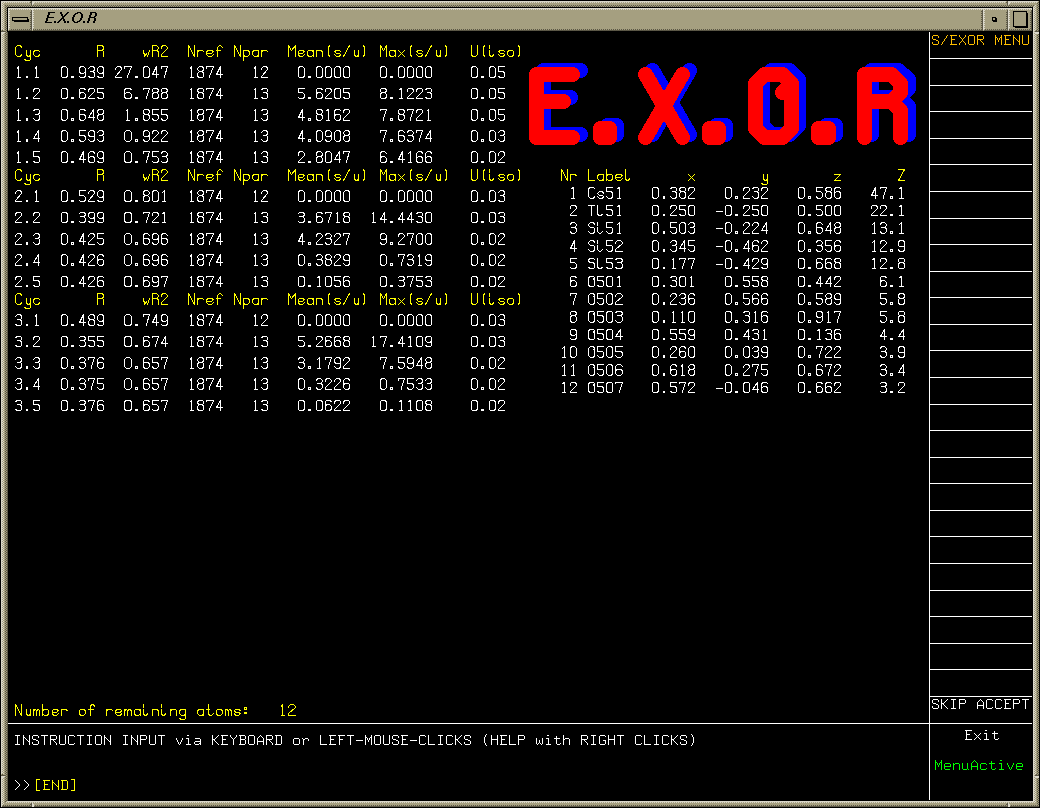
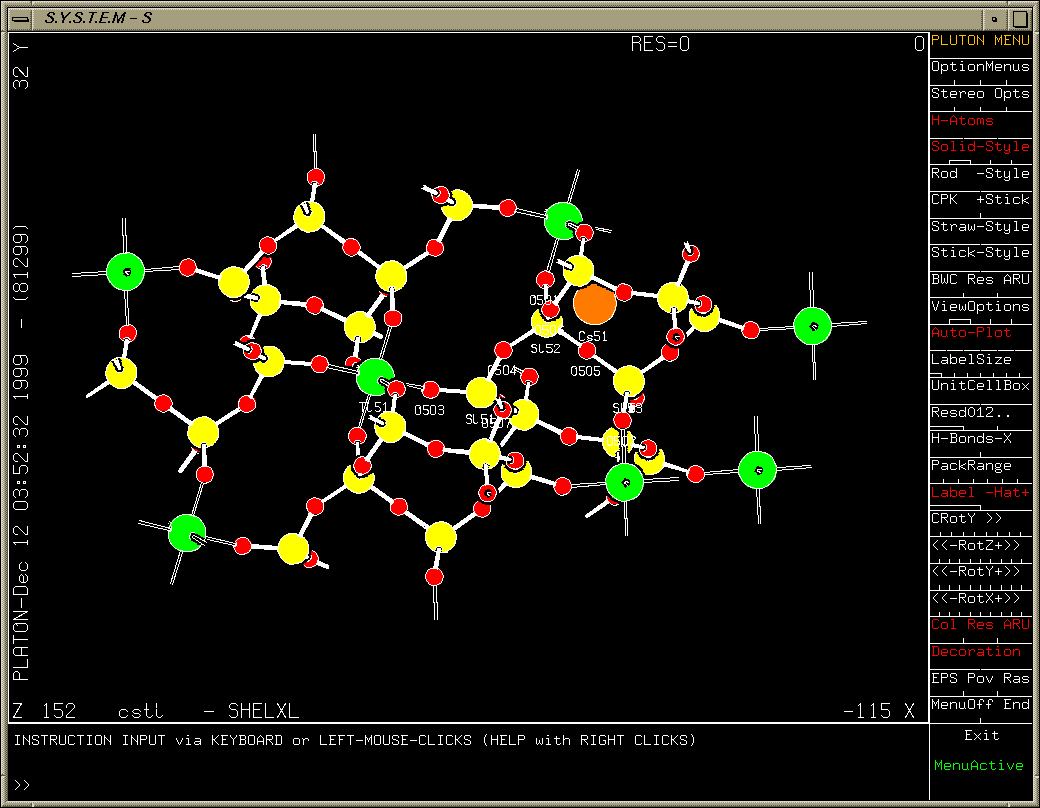
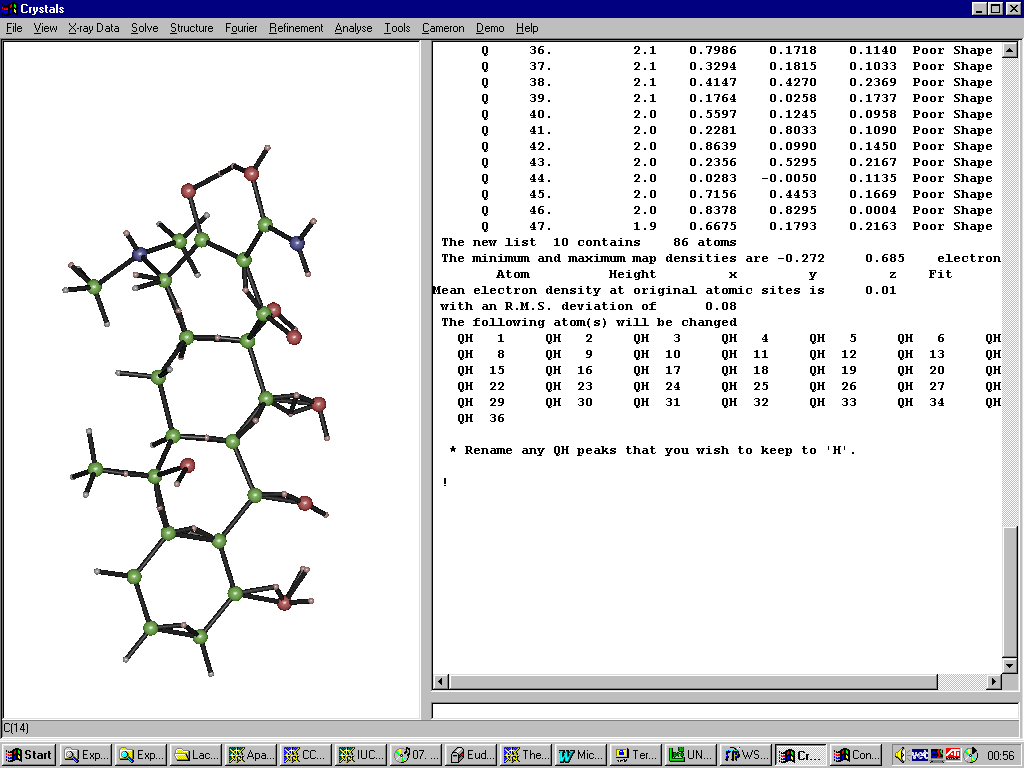
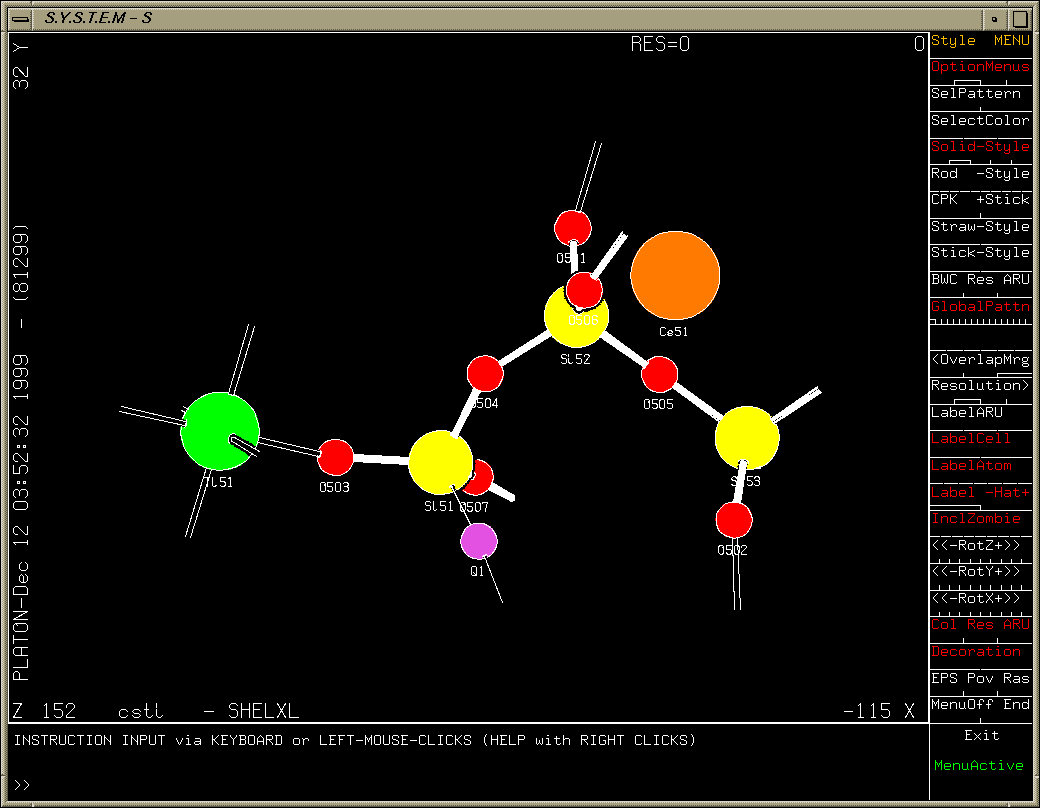
One oxygen atom left to find (after refining heavy atoms anisotropically). Purple Q peak stands out quite clearly.
Graphically Interacting with the Structure
Most freely available programs and suites now have the ability to interact directly with a graphical representation of the structure during solving and refining.
For routine and not so routine refinements, point and click interfaces are now available, complementing the ASCII file or command line interfaces.
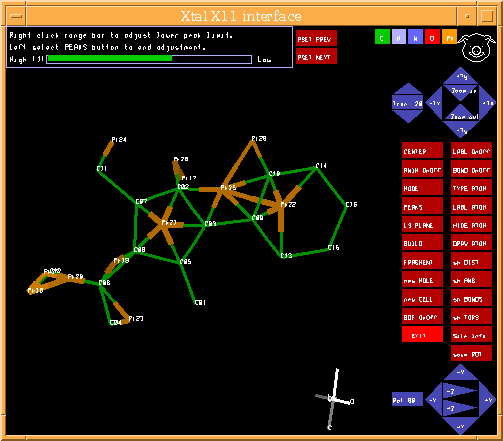
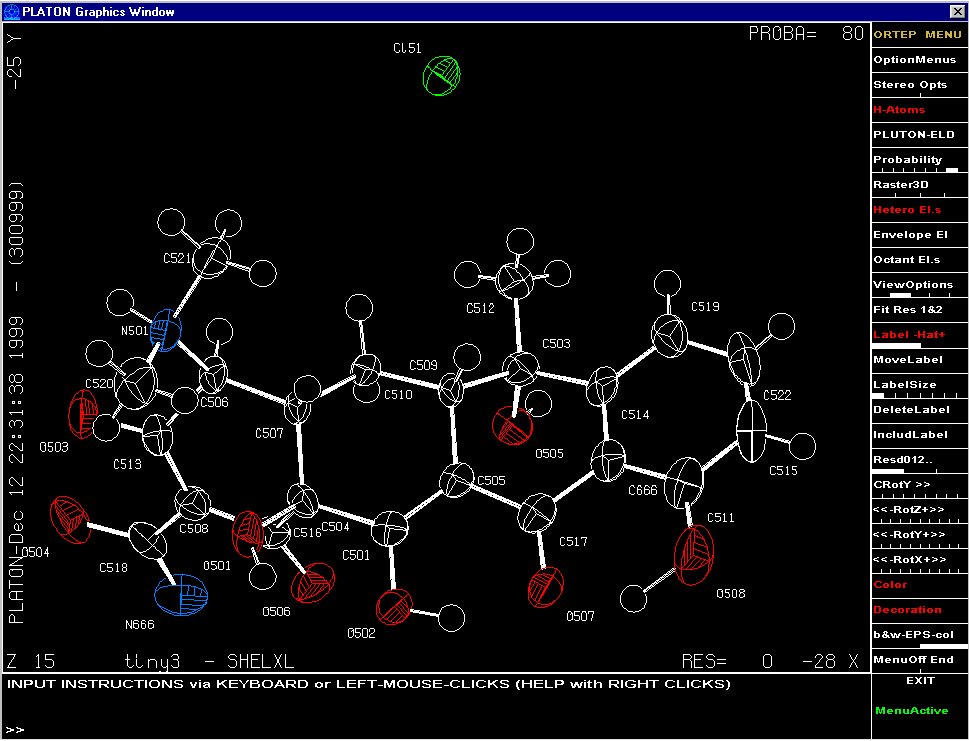
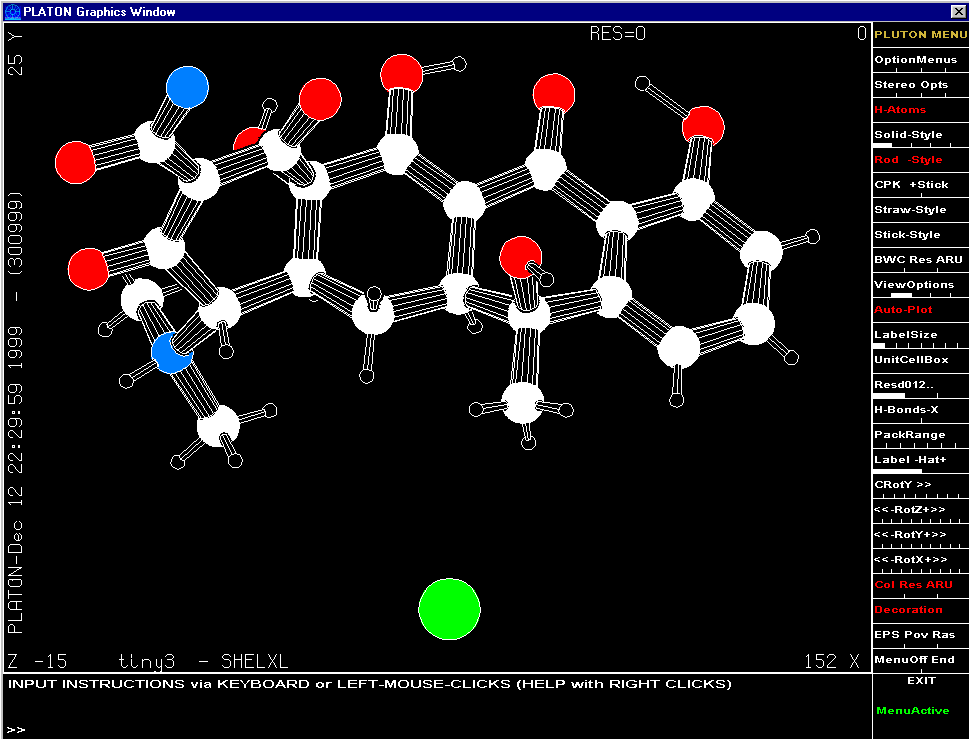
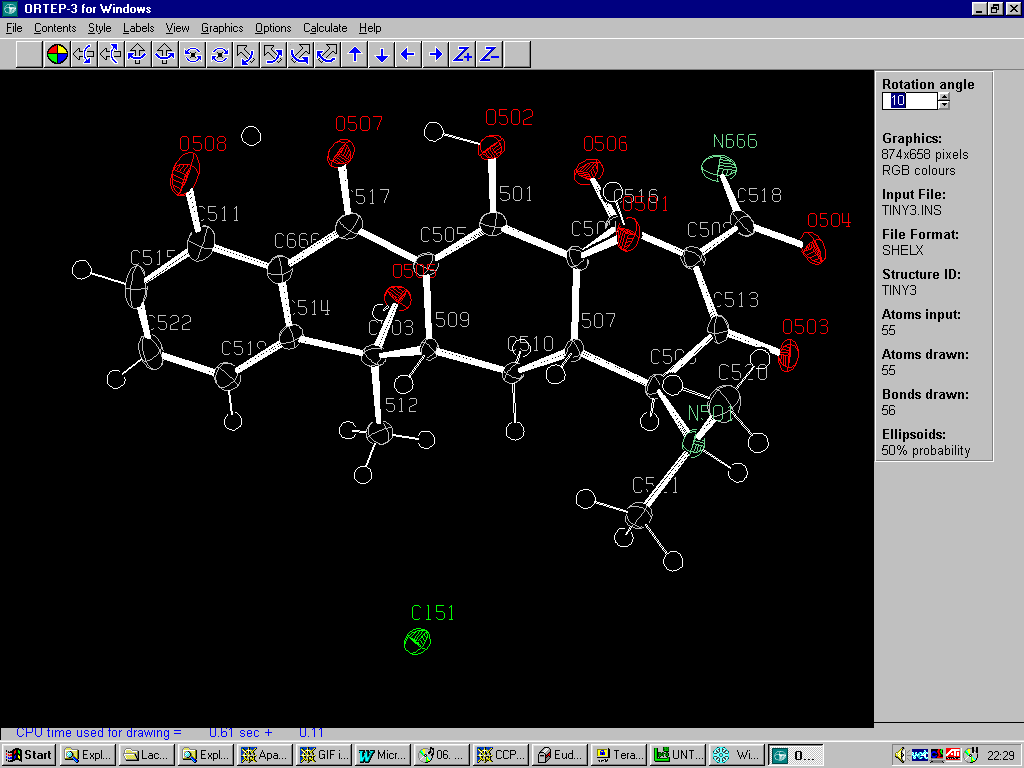
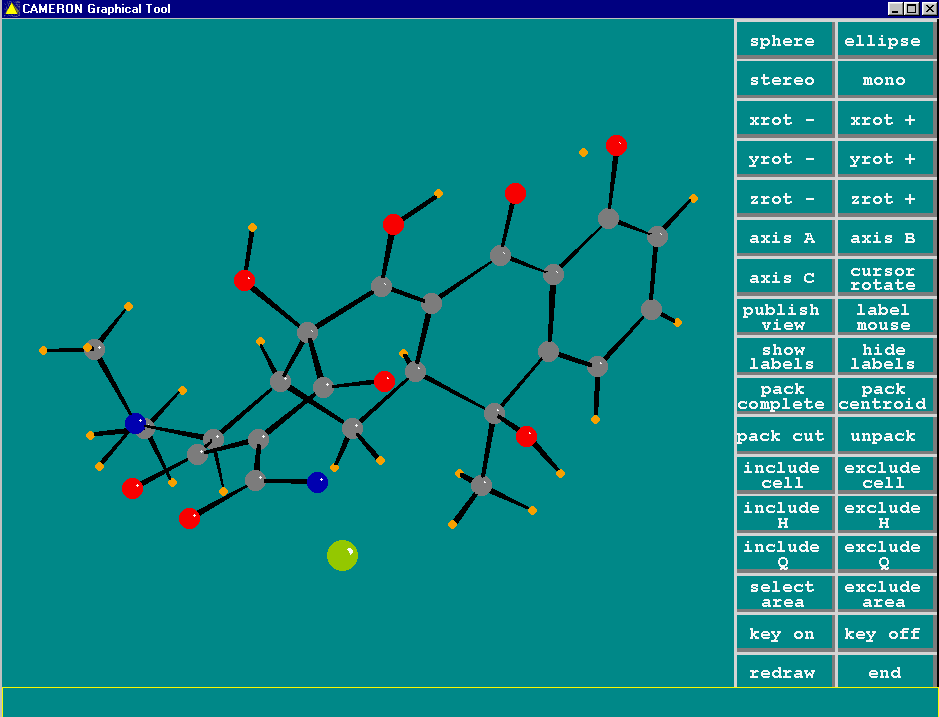
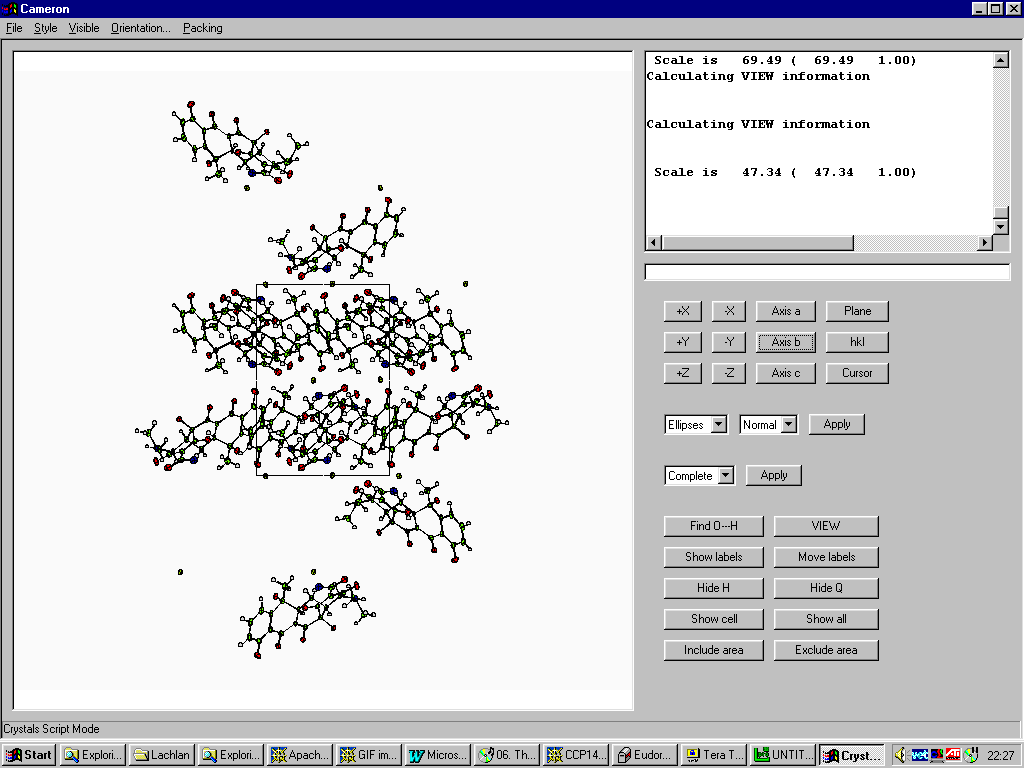
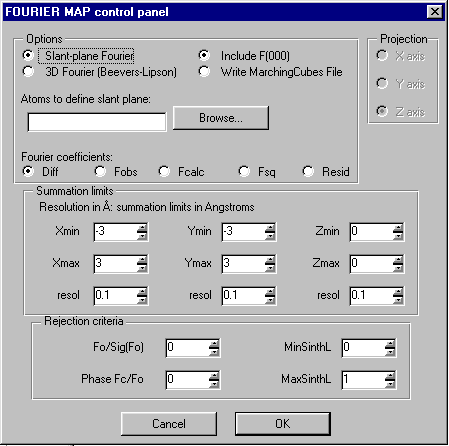
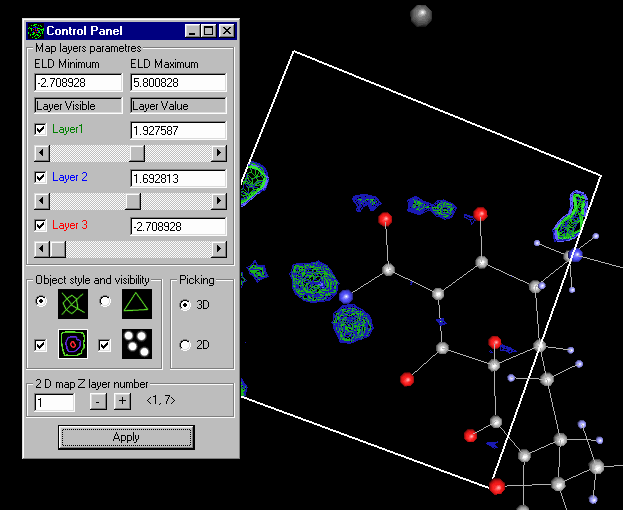
Fourier Map Generation and Viewing
With suites such as WinGX, Platon/System S. Xtal and Crystals, it is trivial to generate Fourier Contour Maps for detailed viewing and analysis.
WinGX:
The WinGX suite has many strong points and one is it’s Fourier Map generation and viewing utilities.
In the above Cesium Titanium Silicate, thermals on one or two of the oxygens seem quite large. If you don’t believe your eyes, a check through Platon will tell you about it.
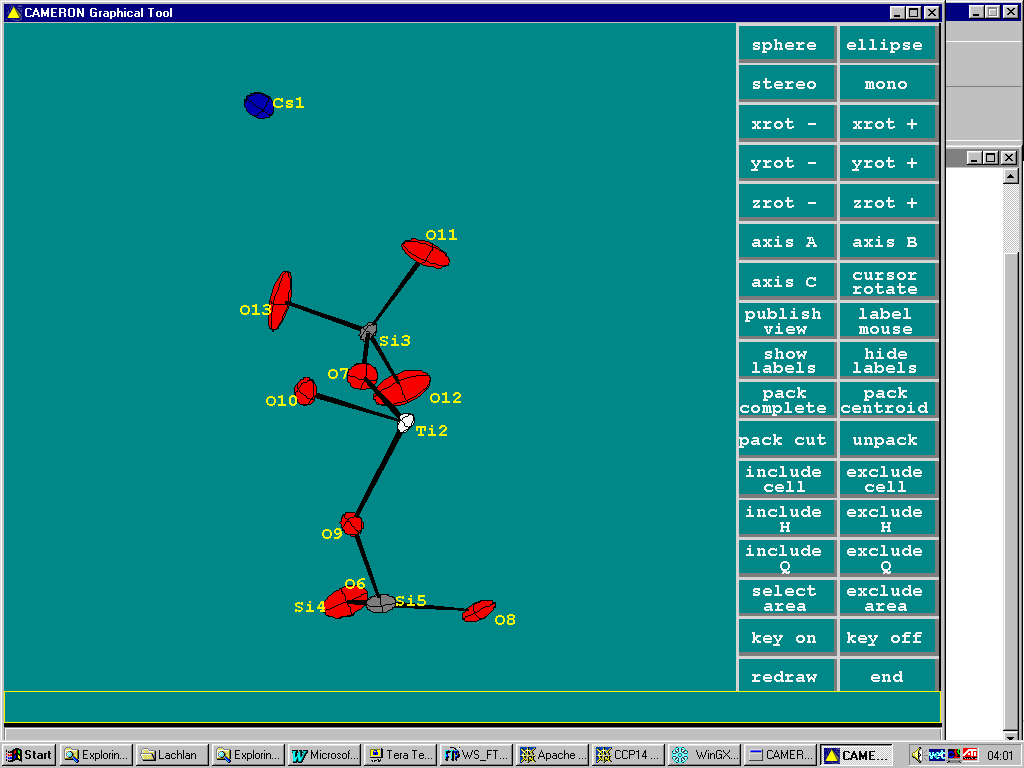
Doing an Shelxl OMIT on the offending atom, go into the WinGX Fourier Map Generator tpo generate a difference map (either slant plane of 3D):
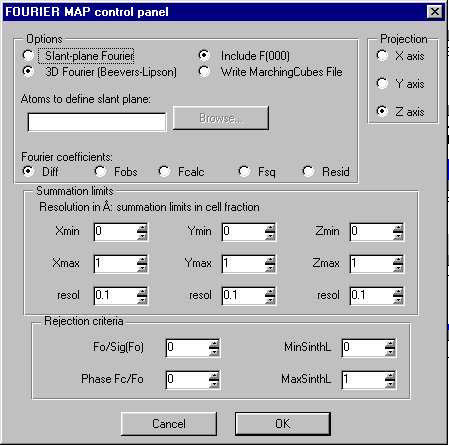
Generate the map:
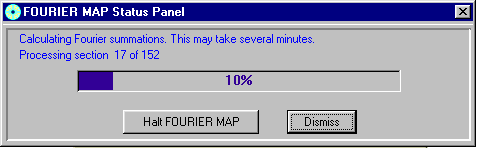
Animating through the map, it is possible to see a slight peanut shape in the difference map of the oxygen showing a slight split atom problem.
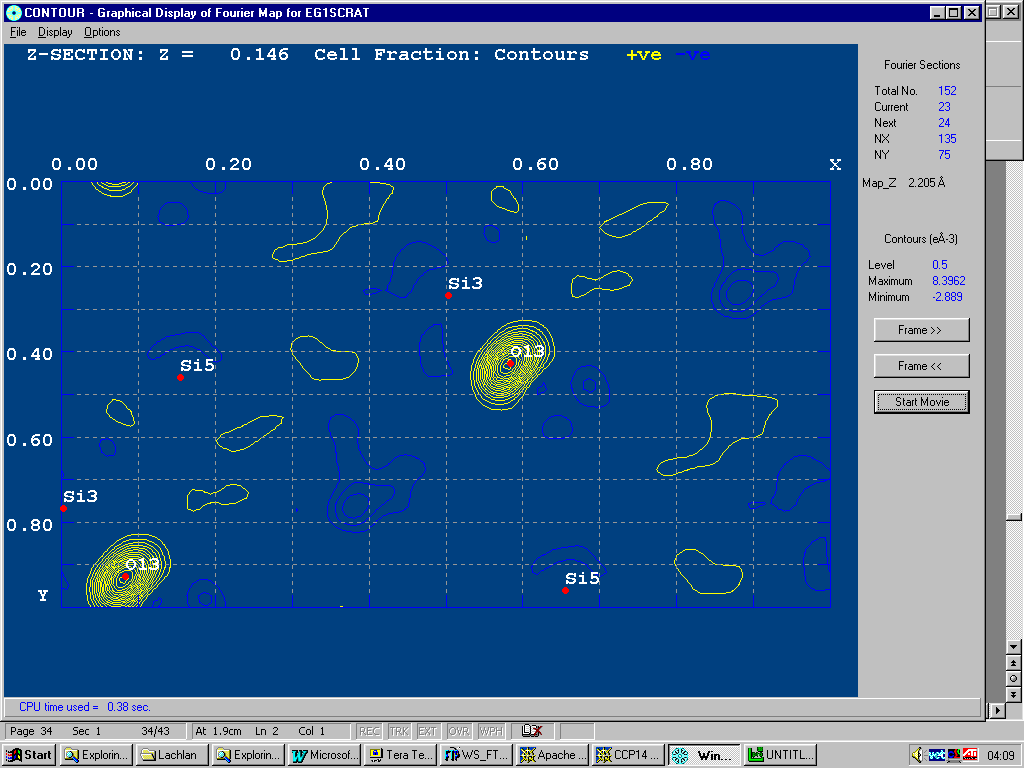
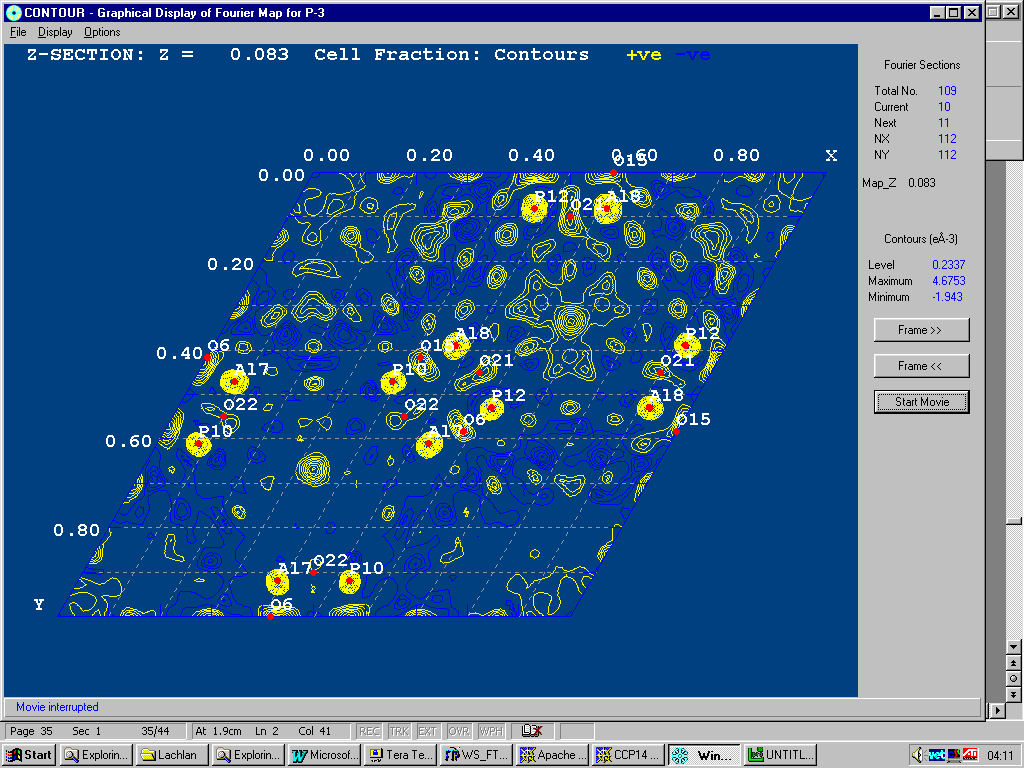
Example of WinGX fourier map animating through a zeolite with disordered template.
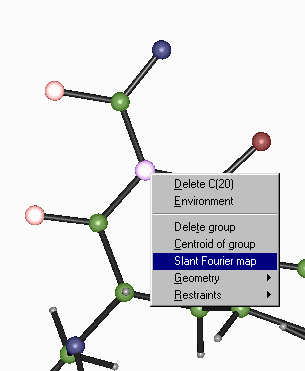
Crystals – Slant Plane Fourier Map:
Define the Slant Plane by selecting 3 atoms then right click to go into the Fourier Map option.
Examine slant plane difference map looking at hydrogen positions – via the Marching Cubes 3D Fourier Map viewer by Michal Husak (also linked into WinGX – and potentially any other program that supports the formats Marching Cubes recognises)
http://mysak.umbr.cas.cz/~husakm/Public/MarchingCubeELD/MarchingCubeELD.htm
http://www.ccp14.ac.uk/ccp/web-mirrors/marchingcube-fourierviewer/~husakm/Public/MarchingCubeELD/MarchingCubeELD.htm
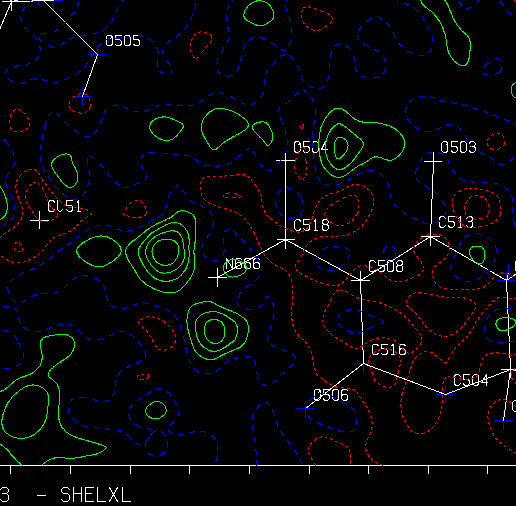
Platon/System S Fourier Map:
Examining slant plane difference map looking at hydrogen positions.
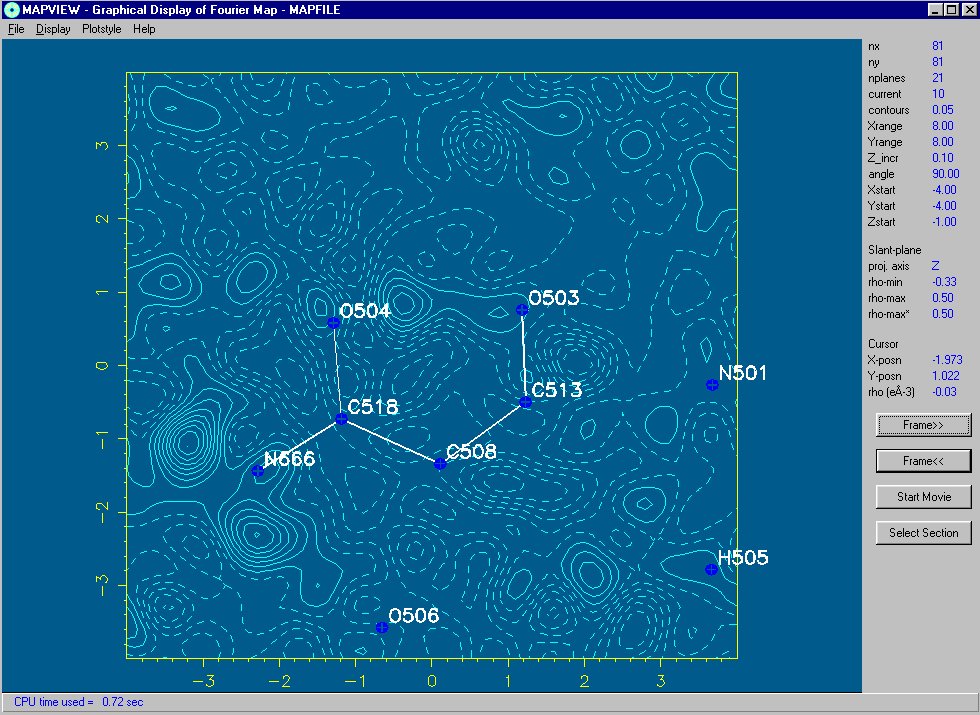
Using WinGX MapView:
(Can be run as a freestanding program linked into any program that will output a Mapview friendly format)
Examining slant plane difference map looking at hydrogen positions
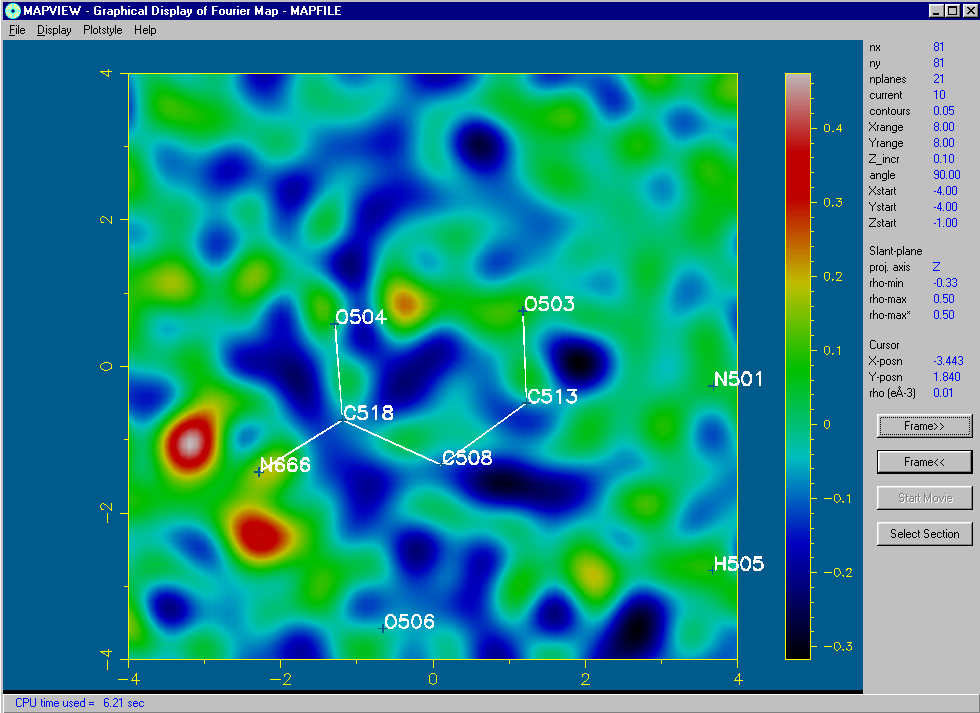
Using the 2D Bitmap Mode of MapView:
Structure Quality Checking
(Each suite can offer different features)
ORTEX:
Example of the Void Finding and graphical viewing within ORTEX (including estimate of time to completion)
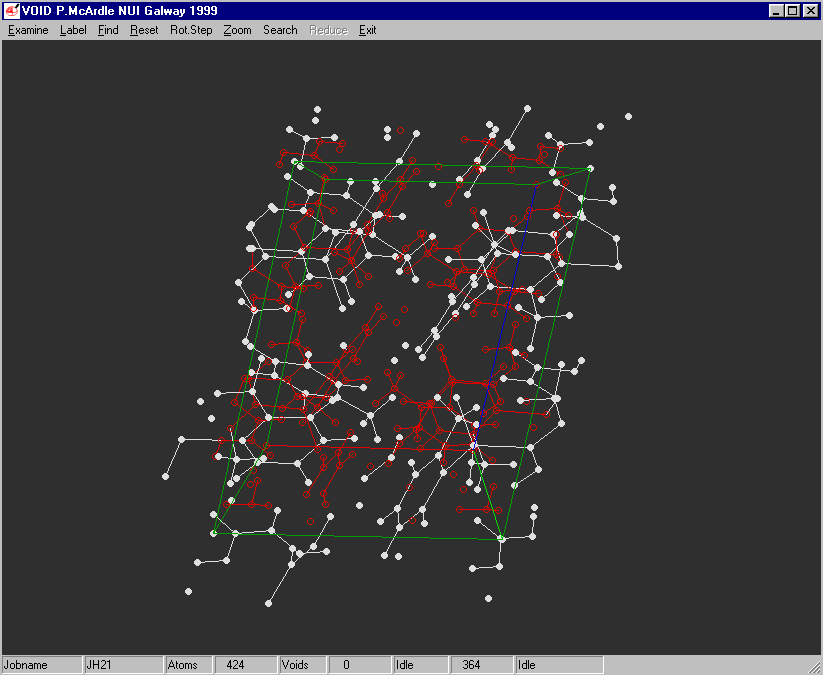
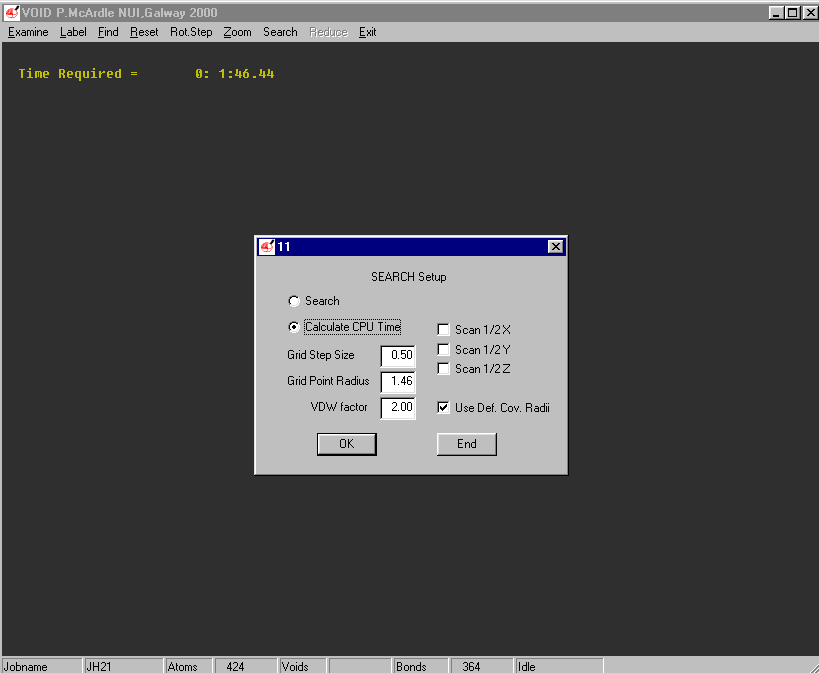
Structure Quality Checking
WinGX:
Links to a wide variety of Structure Checking, Analysis Options and Utilities (including a Windows version of Platon).
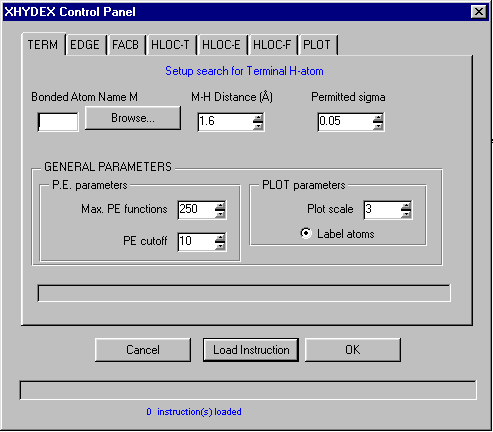
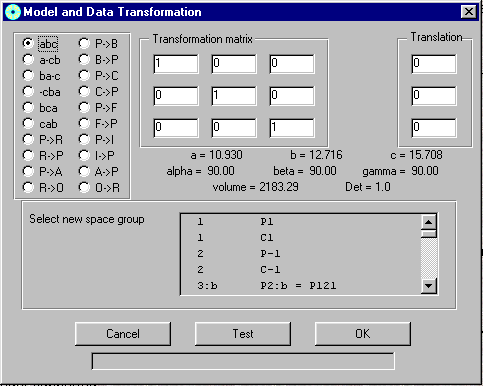
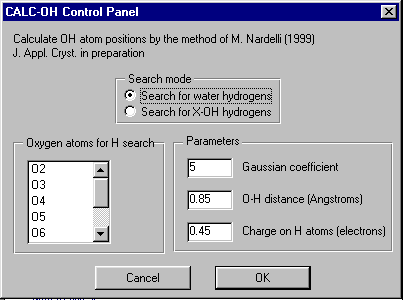
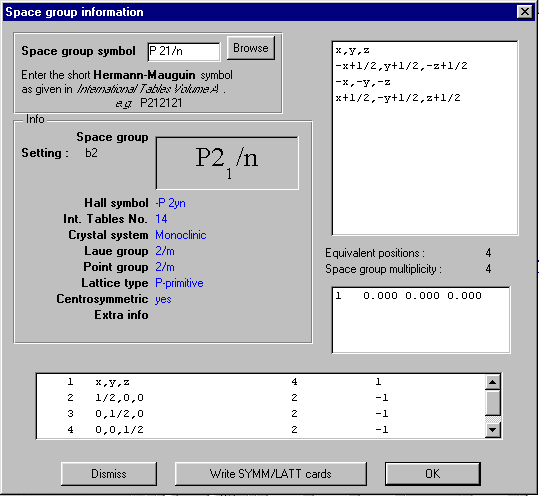
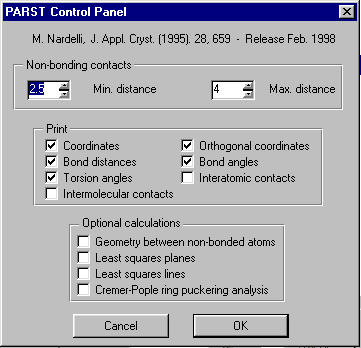
GUI XHYDEX (GUI over code by G. Orpen): Searching for Likely hydrogen positions. GUI CALC-OH (Nardelli 1999) GUI Parst, GUI Model and Data Transformation, SG Info, etc, etc.
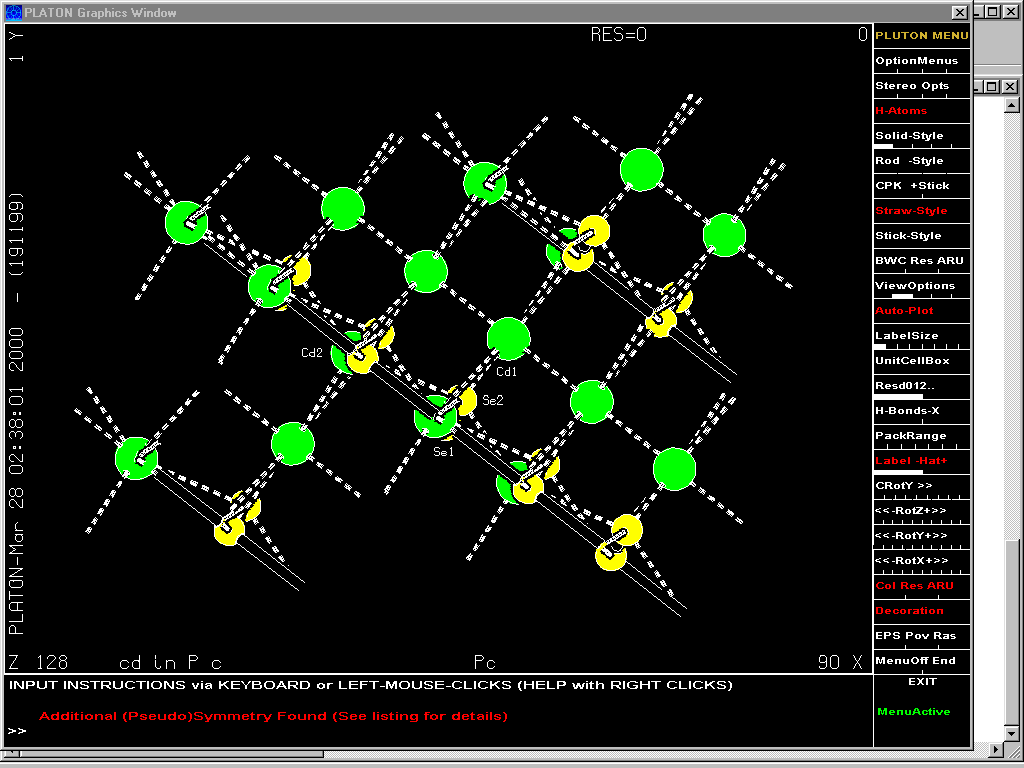
Platon:
Structure checking options such as: Addsym, newman, etc.
Addsym: checking for missing symmetry. Will display or output a modified Shelx file with the updated structure.
Automatic Generation of a Quest Script (and automatically spawn quest if applicable) to see if structure has already been determinated or check out related structures:
Specialist Applications
Charge Density
Project XD
http://www.chem.gla.ac.uk/~paul/paul.html
Anharmonic Refinement
List of Software:
http://www.ccp14.ac.uk/solution/anharmonic/
Incommensurate Structure Refinement
List of Software:
http://www.ccp14.ac.uk/solution/incomm.htm
Exporting/Importing CIF files
Most programs can
export CIF in one form or another.Importing to a useful crystallographic format can be a problem.
Overall refer to IUCr CIF page at:
http://www. iucr.org/iucr-top/cif/
Converting CIF structure file into a Shelx format.
Source Code (and DOS executable): CIF2SX by Louis Farrugia
http://www.chem.gla.ac.uk/~louis/software/
http://www.ccp14.ac.uk/ccp/web-mirrors/farrugia/~louis/software/
Importing both CIF Structure and CIF HKL data for immediate structure analysis and review (have not had chance to put the GNU Xtal suite through its paces):
Windows: WinGX by Louis Farrugia
using the "Model, Import, CIF File" menu option
http://www.chem.gla.ac.uk/~louis/software/
http://www.ccp14.ac.uk/ccp/web-mirrors/farrugia/~louis/software/
UNIX: Platon/System S by Ton Spek
using the "s filename.fcf" command line.
http://www.cryst.chem.uu.nl/platon/
http://www.ccp14.ac.uk/ccp/web-mirrors/platon-spek/platon/
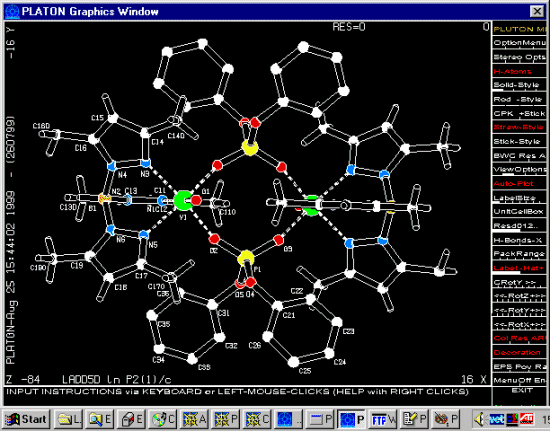
Immediate Structure Visualisation of CIF structure files:
UNIX: Platon/Pluton – Ton Spek
Windows: Ortep-3 – Louis Farrugia (or WinPlaton)
Structure Visualisation and Photorealistic Hardcopy Output
Examples of Software that can do this:
(problem can be that or having a high enough quality printer)
ORTEX (Rendoring and Animation):
http://www.nuigalway.ie/cryst/
http://www.ccp14.ac.uk/ccp/web-mirrors/ortex/cryst/
Just open up a Shelx format *.INS/*.RES file and go for it
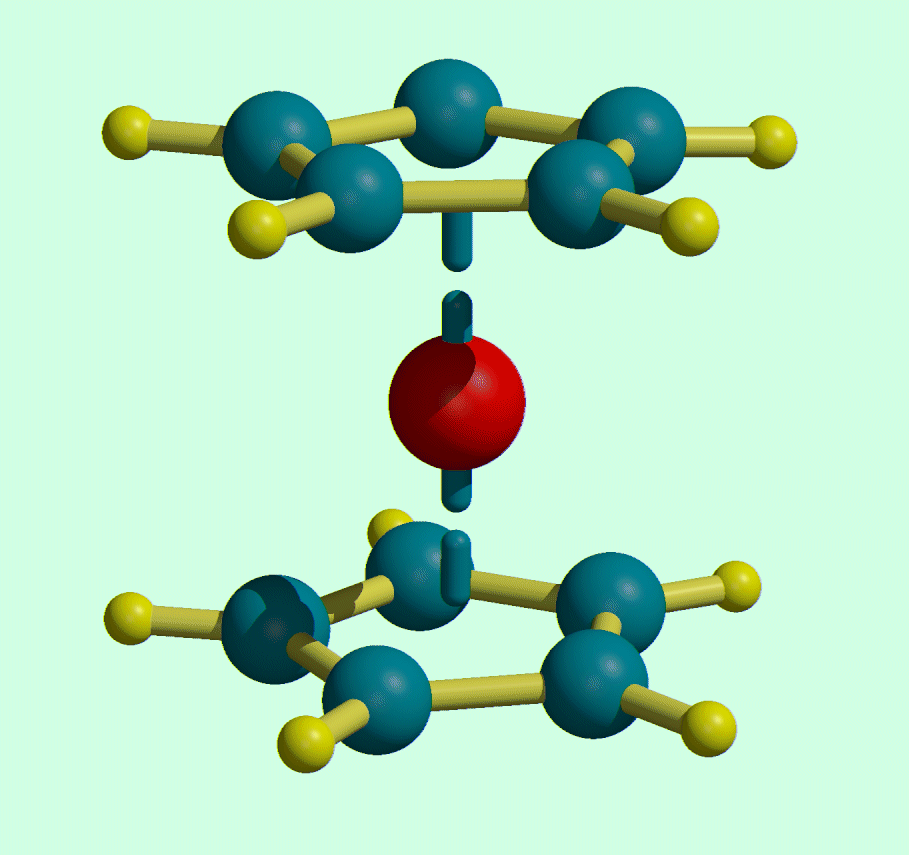
GUI WinOrtep 3/GUI WinStruplo/WinGX
(GUI WinOrtep 3/GUI WinStruplo can be used freestanding)http://www.chem.gla.ac.uk/~louis/software/
http://www.ccp14.ac.uk/ccp/web-mirrors/farrugia/~louis/software/
Can load a variety of structure formats including some Rietveld formats such as GSAS, LHPM, Fullprof, Rietan.
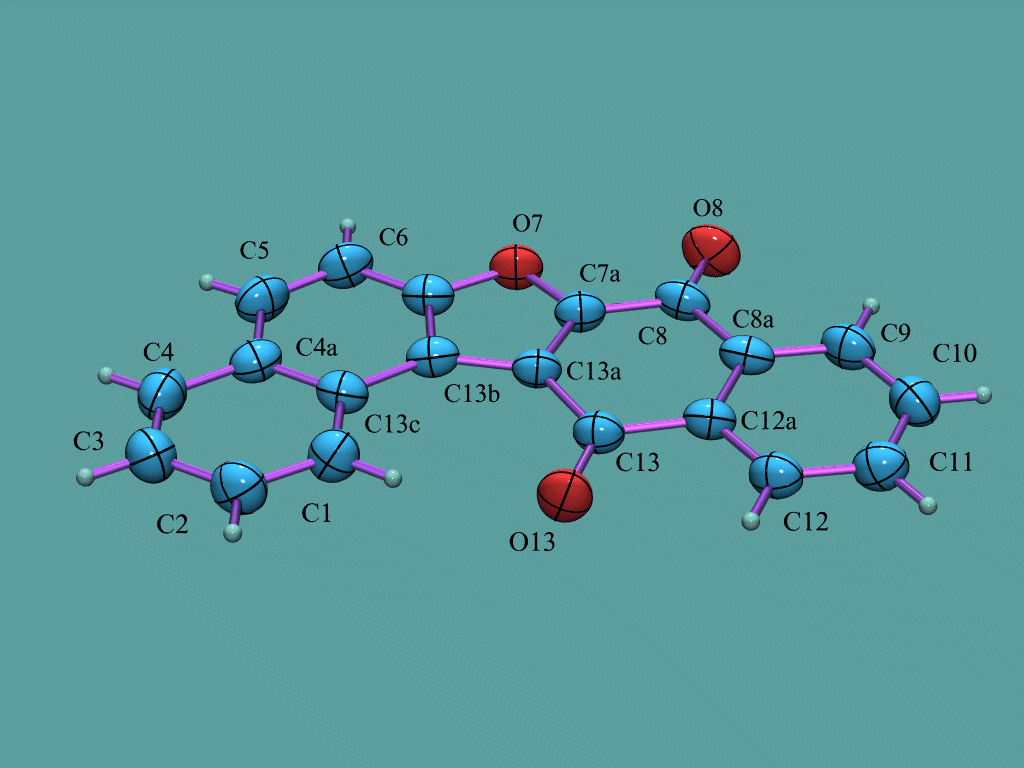
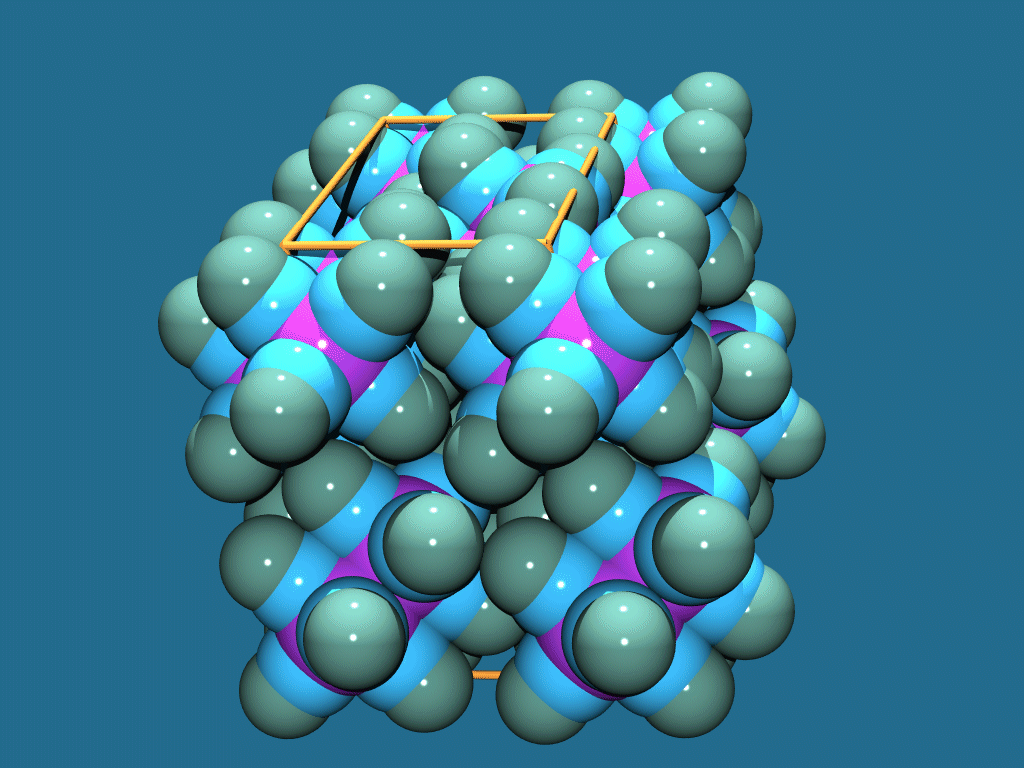
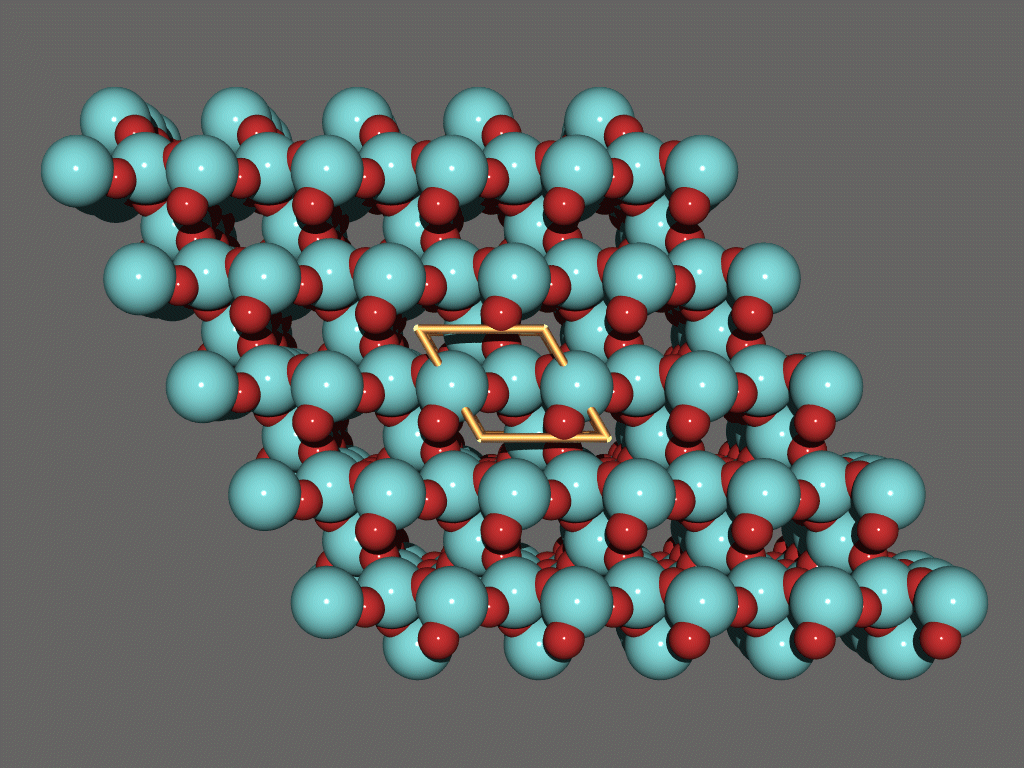

Platon
http://www.cryst.chem.uu.nl/platon/
http://www.ccp14.ac.uk/ccp/web-mirrors/platon-spek/platon/
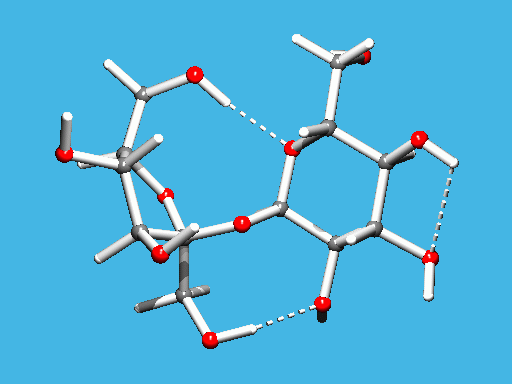
GNU Xtal Suite
http://xtal.crystal.uwa.edu.au/
Dual Boot UNIX/Windows PCs
Get the ability to run a very wide range of crystallographic software on cheap, powerful commodity PCs.
Easy to say this is easy after spending a month or so working it out.
CCP14 based tutorials on installing multi-boot
A local UNIX advocate would be quite happy to get a dual boot system going for you.
Free UNIX Operating Systems (install off the internet)
Linux
refer: http://www.ccp14.ac.uk/solution/linux/
FreeBSD (can run linux binaries)
refer: http://www.ccp14.ac.uk/solution/bsdunix/
(be careful of hackers invading your systems when running Linux/UNIX.
CCP14 tutorials try to be security conscious)VMWARE
(simultaneously run of Windows and Linux):http://www.vmware.com/
(Commercial software)
Freely Available Compilers, GUI Building Kits and Source Code
http://www.ccp14.ac.uk/recomm/
Solving Structures Using Powder Diffraction Data
Highly non-trivial compared to single crystal.
You may be lucky to get beyond correctly indexing the powder data and obtaining a cell.
You might also find that Powder Diffraction Rietveld Structure Refinement software is around 5 to 20 years behind in usability, interaction and robustness compared to single Crystal Equivalents such as Shelx, Crystals, CAOS, Sir97, GNU Xtal and the Single Crystal Suites.
This is not to say that Rietveld refinement programs are not any good. It is mainly an indicator on how powerful modern single crystal programs have become in dealing with large and complex structures.
Solving Structures Using Powder Diffraction Data
Some Links
Powder Data Conversion:
http://www.ccp14.ac.uk/solution/powderdataconv
Search-Match/Phase Identification:
http://www.ccp14.ac.uk/solution/search-match.htm
Powder Indexing
http://www.ccp14.ac.uk/solution/indexing/
Unit-cell Refinement:
http://www.ccp14.ac.uk/solution/unitcellrefine/
LeBail Method for Intensity Extraction
http://www.ccp14.ac.uk/solution/lebail/
Pawley Method for Intensity Extraction
http://www.ccp14.ac.uk/solution/pawley/
Powder Diffraction Structure Solution Pathways
http://www.ccp14.ac.uk/solution/powder_structure_solution_pathways/
Available Rietveld Software
http://www.ccp14.ac.uk/poster-talks/rietveld-csiro-axaa-99/
If you see too many software options, feel free to E-mail the CCP14 (ccp14@dl.ac.uk). Pros and cons of various software can depend on the structure type.
Example Powder Indexing and Spacegroup Assignment
"Crysfire" Indexing Suite
(ito, treor, dicvol, taup, lzon, fjzn, losh and kohl)
http://www.ccp14.ac.uk/tutorial/crys/
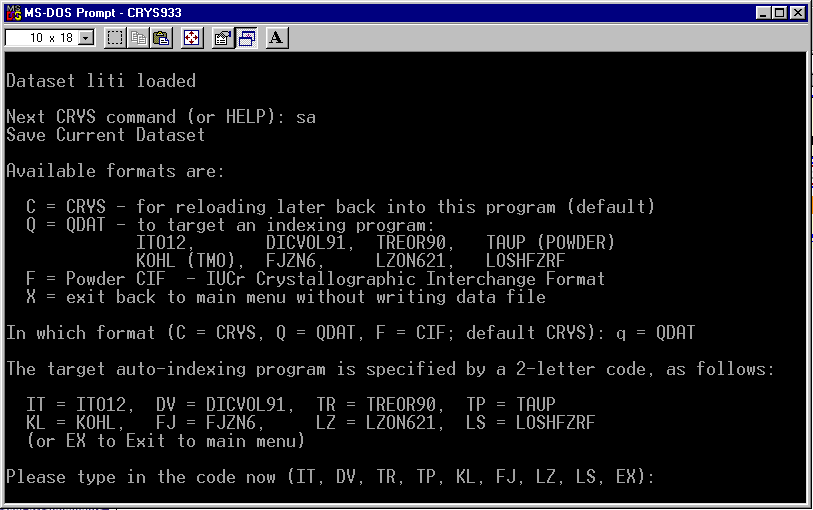
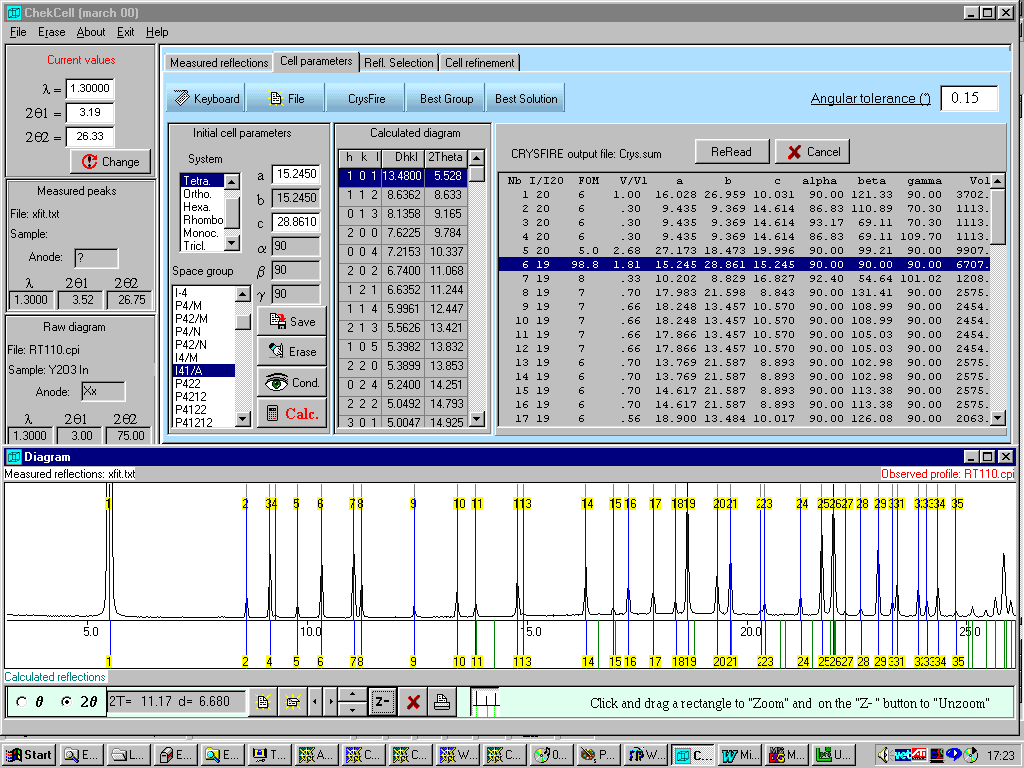
Chekcell
for "automatic or manual" checking cells and assigning Spacegroups(Reads a CRYSFIRE *.SUM summary file)
http://www.ccp14.ac.uk/tutorial/lmgp/
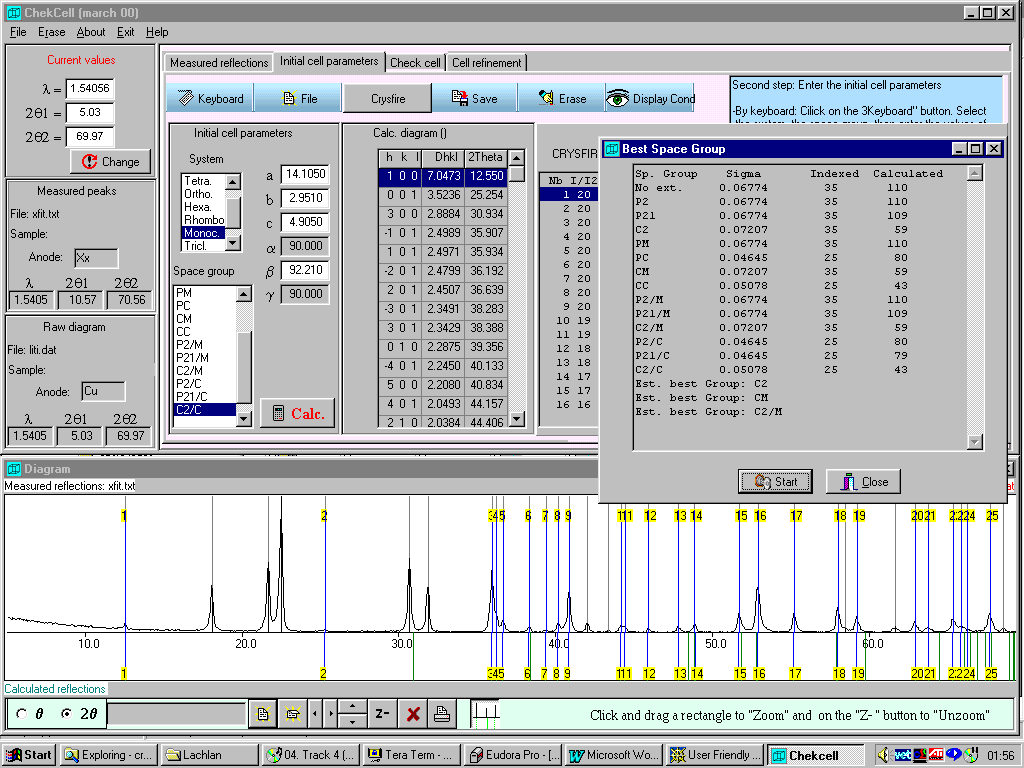
Example of Unsolvable cell on powder data made routine by not relying on "favourite" programs.
(using
May 30th 2000 beta version of Chekcell)Favourite program here was to use Treor by itself and ASCII output file. Highly non-trivial to impossible.
Made Routine by combination of CRYSFIRE (incorporating Treor) and Chekcell – despite reporting of the cell by Ito indexing program as orthorhombic a=c
Tetragonal: a=15.2450, c=28.8610 (I41/AMD or I41/A; Volume=6707)
One spurious peak and possible default volume restrictions used in favourite stand-alone Treor.
Example Structure Solving from Powder Diffraction
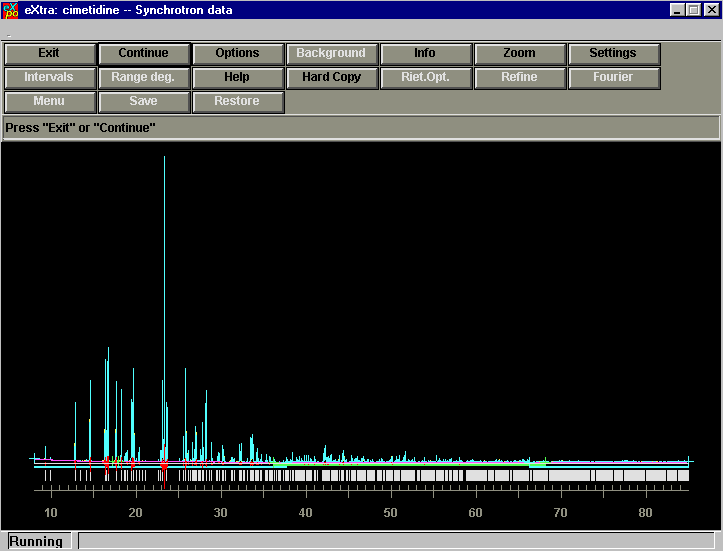
"EXPO"
Direct Methods Software from the Sirware Group(sequel to Sirpow – Sir97 for Powder Diffraction)
http://www.ba.cnr.it/IRMEC/SirWare_main.html
http://www.ccp14.ac.uk/ccp/web-mirrors/sirware/IRMEC/SirWare_main.html
"Program of First Choice"
Downloading of Software
The above software and resources can be downloaded via the CCP14 site:
http://www.ccp14.ac.uk
If any queries, E-mail:
ccp14@dl.ac.uk
or:
L.Cranswick@dl.ac.uk
Graphical tutorial run-throughs of most of this software is located via ("look before you try"):
http://www.ccp14.ac.uk/tutorial/
Software is installed on the CCP14 laptop for demonstration at any time during this meeting.
Where allowed, can be installed on any local computer for demonstration.