Russian Journal of Coordination Chemistry, Vol. 21, No. 3, 1995, pp. 153-161. Translated from Koordinatsionnaya Khimiya, Vol. 21, No. 3, 1995, pp. 163-171. Original Russian Text Copyright � 1995 by Serezhkin, Blalov,Shevchenko
Voronoi-Dirichlet Polyhedra for Uranium(VI) Atoms in Oxygen-Containing Compounds
V. N. Serezhkin, V. A. Blatov, and A. P. Shevchenko
Samara State University, Samara, Russia
Received March 14,1994
Abstract - The geometric characteristics of Voronoi-Dirichlet polyhedra (VDP) for 354 U(VI) atoms in the structures of oxygen-containing compounds are established. It is shown that the volumes of VDPs, as distinct from the volumes of the corresponding coordination polyhedra, are virtually independent of the coordination numbers of uranium atoms and are on average 9.18(25) µ3. The results obtained are in agreement with the previously suggested model of deformable spheres describing the packing of structural units in uranium(VI) compounds.
It is known that the data on crystal structures obtained from X-ray or neutron diffraction investigations represent the totality of point coordinates corresponding to the equilibrium positions of atoms in the unit cell. On the basis of this information, what is known as the description of the crystal structure is created. For this purpose, the points corresponding to atomic positions are linked by segments symbolizing chemical bonds between pairs of atoms; in this way, an infinite unoriented graph is obtained. In crystal chemistry, it is conventional to use the local topological characteristics of this graph, in particular, atomic coordination numbers and coordination polyhedra (CP) for its qualitative and quantitative description. In this case, the coordination number of a certain atom A (that is, the order of a certain vertex of the graph) equals the number of segments (that is, A-X bonds) that converge to the point corresponding to the position of the A atom. The question as to precisely which X atoms are linked to the A atom under consideration (i.e., enter into the first coordination sphere) in the structure of a particular crystal is settled in classical crystal chemistry by analyzing interatomic distances, that is, the lengths of the A-Xi segments. All of the atoms (A, X, etc.) are regarded as rigid spheres [1, p. 22] of a particular radius. In the case when the experimentally found r(A-Xi) distance does not exceed the standard r(A-X) value, which equals the sum of the radii of these atoms, the corresponding pair of atoms is considered to be chemically bonded, and the Xi atom is to be included in the coordination sphere of the A atom.
The crystal structure of a particular compound is considered in this approach as a specific three-dimensional periodic packing of an infinite set of spheres. The atoms of each chemical element in this packing correspond to the spheres of a particular radius; the proportion of spheres of different radii is defined by the stoichiometric composition of the compound, and the coordination number of any atom equals the total number of adjacent spheres touching the sphere corresponding to this atom.
Because the determination of interatomic distances by the data of X-ray diffraction analysis is a trivial problem, it is not surprising that the essentially one-dimensional considered method of determination of atomic coordination numbers in the crystal structure has been widely accepted in spite of a number of disadvantages.
On the one hand, the interaction between two atoms (for example, A and X) in the crystal structure may be generally described in terms of strong (ionic, covalent, and metallic) or weak (van der Waals) chemical bonding. Therefore, not a single but at least several values correspond to different types of bonds for atoms of the same chemical nature in the available systems of crystalline radii. Moreover, for a given type of chemical bond (in particular, ionic), a certain atom is characterized by several more values of radii that depend on the oxidation state and spin state of the atom and even on the value of the coordination number desired. Therefore, it is not surprising that, for a particular pair of atoms A and X, the distance r(A-X) may vary within a large range, even when a strong chemical bond occurs between these atoms. For example, the distances between the U(VI) and oxygen atoms in the structures of known uranyl complexes fall in the range from 1.60 to 2.65 µ [2]. Thus, because a given atom can generally simultaneously participate in the formation of chemical bonds of several types, it is theoretically impossible to represent this atom as a sphere of a fixed radius.
On the other hand, the representation of atoms by rigid undeformable spheres, which has played a great role in the formation of crystal chemistry, is now in contradiction to the modem concept of the nature of chemical bonds. Even on the assumption that the electron-density distribution of the isolated A and X atoms has a spheric shape, the formation of a strong chemical bond between these atoms should be considered a result of the overlapping of their outer valence orbitals, which may be considered as an "interpenetration" of atoms [1, p. 87]. Because it is impossible to estimate the extent of this overlapping (interpenetration) on the grounds of crystal structure data, even the simplest and most reliable atomic radii (covalent or metallic), equal by definition to half the shortest interatomic distance in the structures of simple compounds and true only for the model of touching rigid spheres, lose their physical meaning.
Even without additional arguments, the above-mentioned reasoning evidences the conventionality of the representation of atoms in the crystal as rigid spheres of a particular radius; moreover, the electron density distribution at a certain atom loses its spheric shape under the action of the crystal field. In this connection, the concept of atomic radii in the crystal structure is thought to be, to a great extent, arbitrary [1, p. 82].
An alternative "three-dimensional" method based on the use of the Voronoi-Dirichlet polyhedra (VDP) or atomic domains is known [3] in addition to the considered classical "one-dimensional" method of the determination of atomic coordination numbers.
It is known that the VDP of a certain atom is the polyhedron of the smallest volume formed by the planes passing through the middles of and normal to the segments joining this atom with each of its possible neighbors. For example, in the case when an A atom is surrounded in a crystal by six X atoms forming a CP in the form of a regular octahedron or trigonal prism, the VDP of this atom has the form of a cube or a trigonal bipyramid, respectively. Thus, the coordination number of the A atom is generally equal to the number of faces of its VDP, because each face of the VDP corresponds to one vertex of the coordination polyhedron, and the VDP is dual to the CP. Below, the polyhedra obtained by the procedure used for constructing VDPs at an arbitrary value of Kd {Kd is the coefficient of division of the A-X bond, with a perpendicular plane passing through a point Z on the AX segment; this coefficient equals the ratio r(A-Z)/r(A-X)] are called dual CPs. Thus, the VDP is a special case of the dual CP at Kd = 0.5. A specific VDP corresponds to each crystallographic type of atom in the structure of the compound, and the crystal structure as a whole may be considered as a totality of VDPs touching each other with congruent faces, completely filling the entire space of the crystal, because any point of the crystal belongs to at least one VDP. As noted above [4], in the structure of simple compounds, one can regard the VDP as a geometric image of an atom in a certain crystal field and consider it the region of action of this atom, because any point of this area is closer to "its own" atom than to any adjacent one of the same chemical nature.
Although the method of determination of the atomic coordination number in crystals by means of VDPs has been known for a rather long time, as far as we know it has very rarely been used in the crystal chemistry of inorganic and coordination compounds. On the one hand, this is due to the fact that calculation of the geometric characteristics of VDPs is very tedious, especially for crystals of low symmetry containing a large number of crystallographically different atoms. On the other hand, the application of VDPs in the structural analysis of inorganic and coordination compounds is hindered by the fact that the problem of taking into account the distinctions in the chemical nature of atoms arises in the construction of the regions of atomic action, because the problem of the isolation of the spatial region of an atom of each chemical sort has not been solved even for the case when two sorts of atoms are present. It was shown recently [4] that, for structures with several chemical sorts of atoms, the VDP describes the region of action of a certain atom, correct to similarity, under conditions of uniformity of the nearest environment of this atom, that is, the occurrence of only atoms of a single chemical sort in the first coordination sphere of the basis atom. In accordance with [5], the regions of atomic action should be thought of as the basins of nuclei-attractors in the vector field of the electron density gradient.
This work was undertaken with the aim of studying more thoroughly the equality of the VDP volumes of uranium(VI) atoms found in [4] for crystals of 30 compounds. Simultaneously, we planned to verify the possibility of the application of VDPs for the determination of the coordination numbers of U(VI) atoms in the structures of oxygen-containing compounds using a wider variety of objects.
GEOMETRIC CHARACTERISTICS OF THE VORONOI-DIRICHLET POLYHEDRA OF U(VI) ATOMS
The calculation of characteristics of the VDPs of U(VI) atoms in the structures of oxygen-containing compounds, including uranyl complexes and simple and mixed oxides (uranates), was carried out by the structurally topological TOPOS software [6]. These programs make it possible to calculate atomic VDPs in crystal structures of any degree of complexity. The original information on crystal structures contained in the Cambridge crystallographic databank and the databank for uranyl compounds [7] was used in the calculations, provided that three conditions were fulfilled: (1) the crystal structure was determined with Rf
� 0.10; (2) in accordance with the classical description, the UOn polyhedra occur in the structure; and (3) statistically disordered uranium and oxygen atoms are not in the structure. The data on the structure of 276 compounds, which contain 354 crystallographically different sorts of uranium atoms in their structures, satisfied the requirements indicated. Hydrogen atoms exist in 236 of the 276 compounds; however, their coordinates were determined in the structures of only 95 compounds. Considering that the accuracy of the determination of the coordinates of hydrogen atoms in the presence of a uranium atom is low and that the positions of these atoms were not established in the structures of 141 compounds, the hydrogen atoms were disregarded in the calculations of the VDPs of uranium atoms.Table 1. Characteristics of the dual coordination polyhedra of U(VI) atoms at Kd(U-O) � 0.5
|
CP com- position |
N* |
Kd=0.2 |
Kd=0.3 |
Kd=0.4 |
Kd=0.5 |
VCP**, µ3 |
||||
|
VS, µ3 |
SS, µ2 |
VS, µ3 |
SS, µ2 |
VS, µ3 |
SS, µ2 |
VVDP, µ3 |
SVDP, µ2 |
|||
|
UO6 |
28 |
0.59(2) |
4.23(9) |
1.98(6) |
9.5(2) |
4.70(13) |
16.9(3) |
9.2(3) |
26.5(5) |
12.1(4) |
|
UO7 |
209 |
0.58(2) |
4.11(8) |
1.97(5) |
9.2(2) |
4.67(13) |
16.4(3) |
9.1(2) |
25.7(5) |
15.7(4) |
|
UO8 |
110 |
0.60(1) |
4.11(7) |
2.01(5) |
9.2(2) |
4.76(11) |
16.4(3) |
9.3(2) |
25.7(4) |
18.3(5) |
|
UO9 |
1 |
0.62 |
4.24 |
2.09 |
9.5 |
4.95 |
17.0 |
9.7 |
26.5 |
21.8 |
|
UO10 |
5 |
0.61(2) |
4.30(5) |
2.05(6) |
9.7(1) |
4.86(13) |
17.2(2) |
9.5(3) |
26.9(3) |
26.7(3.0) |
|
UO12 |
1 |
0.59 |
4.20 |
1.98 |
9.5 |
4.70 |
16.8 |
9.2 |
26.3 |
29.8 |
|
UOn*** |
354 |
0.59(2) |
4.12(8) |
1.98(6) |
9.3(2) |
4.70(13) |
16.5(3) |
9.2(3) |
25.8(5) |
16.4(2.3) |
*N is the number of crystallographic sorts of uranium atoms with the dual CP of the corresponding chemical composition.
** The mean value of volumes (µ3) of the classical CPs of uranium atoms, the composition and number of which are indicated in the first and second columns, respectively, of this table.
***This line contains the mean values of characteristics of all of 354 crystallographic sorts of uranium atoms regardless of their coordination numbers.
Because the dependence of the geometric characteristics of dual CPs on Kd has not been studied so far, we First calculated the dual CPs of uranium atoms in the structures of all 276 U(VI) compounds at different values of Kd for the U-O bonds (Kd = 0.5 was used for the other possible bonds). Note in this connection that, in the known systems of crystalline radii, the minimum and maximum radii of the uranium atom (rU) are 1.775 and 0.59 µ, and those of the oxygen atom (rO) are 1.52 and 0.45 µ, respectively [1]. Using these values, one can establish the limiting values of the coefficient of division of the U-O bond by the plane passing normally to this bond and cutting it into segments; the ratio of the lengths of these segments equals the ratio between any known radii of uranium and oxygen atoms. Under the indicated condition, Kd = rU/(rU + rO); therefore, the maximum and minimum possible values are Kd = 1.775/(1.775 + 0.45) = 0.80 and 0.59/(0.59 + 1.52) = 0.28, respectively.
In accordance with the data obtained (Table 1), the dual CPs of uranium atoms have from 6 to 12 faces at any Kd(U-O) < 0.5. For a particular compound, the variation of Kd(U-O) within the range 0 < Kd(U-O) < 0.5 affects only the absolute values of the volume (VS) and total area of faces (SS) of dual CPs. The dual CPs themselves are similar to each other, with the coefficient of similarity being equal to the ratio between the corresponding values of Kd, and thus have identical combinatorial and topological characteristics. Because each face corresponds to a single U-O bond, the CPs of uranium atoms have composition UOn, where n= 6, 1, 8, 9, 10, or 12 at Kd(U-O) < 0.5 (Table 1). As an example. Table 2 gives the results of calculation of the VDPs of uranium atoms for the structures NaCa3UO2(CO3)3SO4F
Ç l0H2O [8] and K2UO2(SO4)3 [9], in which the uranium atoms have coordination numbers 8 and 7.Note that, in accordance with the classical method of determination of coordination numbers considered above, the coordination numbers of U(VI) atoms in oxygen-containing compounds vary from 6 to 8 [2]. In this case, the atomic CPs have the form of a tetragonal, pentagonal, or hexagonal bipyramid. The results that we obtained testify that U atoms, as expected, have dual CPs in the form of a tetragonal, pentagonal, or hexagonal prism at Kd(U-O)
� 0.5 in most of the compounds studied; that is, in 347 cases out of 354, the coordination numbers of the U atoms equal 6, 7, or 8, respectively, as in the classical approach.The results obtained (Table 1) are also indicative of the occurrence of six compounds, in the structure of which seven crystallographically different sorts of uranium atoms display abnormally high coordination numbers (in comparison with the classical values): 9, 10 (in five cases), and 12. According to available data (as an example, Table 3 presents the results of the calculation of VDPs of uranium atoms in two "abnormal" compounds, namely,
b-CdUO4 [10] and UO2(C4H4O5) [11]), an increase in the coordination number is due to the occurrence of two, four, or six U� O contacts with interatomic distances (from 3.12 to 3.83 µ) approximately 1.5-2 times longer than those typical for U(VI) [2]. VDPs in the form of a distorted tetragonal or pentagonal prism with several additional small faces correspond to all of the abnormal coordination numbers (9, 10, or 12) of uranium atoms; the VDPs of uranium atoms corresponding to coordination number 9 = 7 + 2, 10 =6+ 4, and 12=6+6 have two, four, and six additional faces of this type, respectively. The faces of dual CPs corresponding to such contacts (Table 3) have areas that do not exceed 3% of SS. The smallest solidTable 2. Results of the calculations of the VDPs of uranium atoms in the structures of uranium(VI) compounds*
|
Atom X |
Coordinates of the X atom |
r(U-X), µ |
Kd(U-O) |
|||||||||
|
0.5 |
0.6 |
0.7 |
0.8 |
|||||||||
|
x |
y |
z |
Sseg |
W |
Sseg |
W |
Sseg |
W |
Sseg |
W |
||
|
NaCa3UO2(CO3)3SO4F Ç l0H2O [central atom U(l): x = 0.301, y = 0.638, z = 0.189] |
||||||||||||
|
O(2) |
0.312 |
0.648 |
0.312 |
1.77 |
19.7 |
21.3 |
19.5 |
20.9 |
18.2 |
19.4 |
15.8 |
17.0 |
|
O(1) |
0.290 |
0.632 |
0.063 |
1.81 |
20.7 |
21.3 |
20.8 |
21.1 |
20.3 |
20.1 |
19.8 |
18.5 |
|
O(7) |
0.587 |
0.745 |
0.194 |
2.40 |
10.1 |
9.8 |
7.8 |
7.5 |
5.0 |
4.3 |
0.8 |
0.6 |
|
O(10) |
0.121 |
0.351 |
0.192 |
2.42 |
10.1 |
9.7 |
7.8 |
7.4 |
4.9 |
4.2 |
0.8 |
0.5 |
|
O(4) |
0.193 |
0.817 |
0.189 |
2.42 |
9.9 |
9.6 |
7.6 |
7.2 |
4.7 |
4.0 |
0.7 |
0.5 |
|
O(9) |
0.376 |
0.432 |
0.190 |
2.43 |
9.9 |
9.5 |
7.6 |
7.2 |
4.7 |
3.9 |
0.7 |
0.5 |
|
O(6) |
0.508 |
0.921 |
0.192 |
2.43 |
9.7 |
9.4 |
7.4 |
7.0 |
4.6 |
3.9 |
0.6 |
0.4 |
|
O(3) |
0.018 |
0.561 |
0.191 |
2.44 |
9.8 |
9.4 |
7.5 |
7.0 |
4.7 |
3.9 |
0.7 |
0.5 |
|
O(2) |
0.628 |
0.896 |
0.201 |
2.86 |
4.6 |
4.9 |
10.1 |
11.5 |
15.3 |
16.5 |
||
|
C(l) |
0.044 |
0.707 |
0.196 |
2.87 |
4.7 |
5.0 |
10.3 |
11.6 |
15.3 |
16.5 |
||
|
C(3) |
0.232 |
0.312 |
0.196 |
2.87 |
4.7 |
4.9 |
10.3 |
11.6 |
15.5 |
16.6 |
||
|
Ca(l) |
0.660 |
0.548 |
0.226 |
3.99 |
0.7 |
0.6 |
4.6 |
4.0 |
||||
|
Ca(3) |
0.398 |
1.093 |
0.220 |
4.00 |
0.7 |
0.6 |
4.7 |
4.0 |
||||
|
Ca(2) |
-0.147 |
0.282 |
0.226 |
4.00 |
0.7 |
0.6 |
4.7 |
4.0 |
||||
|
K2UO2(SO4)3 [central atom U(l): x = 0.268, y = 0.637, z = 0.501] |
||||||||||||
|
O(15) |
0.160 |
0.588 |
0.457 |
1.854 |
18.9 |
20.8 |
19.0 |
20.7 |
17.6 |
19.4 |
15.2 |
16.6 |
|
O(16) |
0.384 |
0.687 |
0.535 |
1.937 |
20.2 |
20.5 |
20.1 |
20.4 |
18.0 |
18.7 |
14.5 |
15.4 |
|
O(11) |
0.156 |
0.696 |
0.627 |
2.325 |
13.7 |
13.5 |
13.8 |
13.5 |
13.0 |
12.5 |
5.7 |
5.5 |
|
O(14) |
0.209 |
0.698 |
0.319 |
2.348 |
13.3 |
13.1 |
13.4 |
13.1 |
11.3 |
11.1 |
4.8 |
4.3 |
|
O(6) |
0.266 |
0.596 |
0.742 |
2.452 |
12.8 |
12.1 |
12.9 |
12.1 |
9.3 |
9.1 |
3.0 |
2.8 |
|
O(2) |
0.391 |
0.553 |
0.506 |
2.525 |
10.7 |
10.2 |
8.4 |
8.1 |
4.6 |
4.3 |
1.2 |
1.1 |
|
O(3) |
0.360 |
0.600 |
0.279 |
2.553 |
10.3 |
9.8 |
8.2 |
7.8 |
5.1 |
4.5 |
2.0 |
1.5 |
|
S(l) |
0.424 |
0.557 |
0.357 |
3.069 |
4.1 |
4.3 |
9.1 |
10.5 |
12.8 |
14.5 |
||
|
S(4) |
0.229 |
0.750 |
0.237 |
3.643 |
2.1 |
1.7 |
8.0 |
7.7 |
||||
|
S(3) |
0.095 |
0.750 |
0.641 |
3.701 |
0.6 |
0.4 |
6.3 |
6.1 |
||||
|
S(2) |
0.321 |
0.598 |
0.879 |
3.724 |
3.1 |
2.5 |
8.2 |
7.9 |
||||
|
K(5) |
-0.007 |
0.672 |
0.345 |
3.959 |
0.1 |
0.1 |
1.3 |
1.2 |
3.3 |
3.3 |
||
|
K(3) |
0.561 |
0.626 |
0.640 |
4.051 |
<0.1 |
<0.1 |
1.1 |
0.9 |
2.3 |
2.2 |
||
|
K(2) |
0.211 |
0.527 |
0.169 |
4.092 |
0.8 |
0.7 |
2.6 |
2.3 |
||||
|
K(2) |
0.289 |
0.473 |
0.669 |
4.125 |
0.1 |
<0.1 |
1.5 |
1.3 |
3.9 |
3.6 |
||
|
K(3) |
0.061 |
0.626 |
0.860 |
4.323 |
0.5 |
0.4 |
2.7 |
2.4 |
||||
|
K(4) |
0.318 |
0.750 |
0.877 |
4.444 |
0.6 |
0.5 |
1.9 |
1.6 |
||||
|
K(5) |
0.493 |
0.672 |
0.155 |
4.454 |
0.4 |
0.3 |
1.6 |
1.4 |
||||
* In the r(U-X) column, the distance is shown between the central uranium atom and the atom whose designation and coordinates are given in the corresponding line of the table: SSEG is the value of the area of the dual CP face common to the central atom and the atom shown in the line, expressed in % of the total area;
W is the solid angle of the dual CP face common to the central atom and the atom shown in the line, expressed in % of the total solid angle 4p steradian of the uranium atom.Table 3. Results of the calculation of the VDPs of uranium atoms in the structures of "abnormal" uranium(VI) compounds*
|
Atom X |
Coordinates of the X atom |
r(U-X) µ |
Kd(U-O) |
|||||||||
|
0.5 |
0.6 |
0.7 |
0.8 |
|||||||||
|
x |
y |
z |
Sseg |
W |
Sseg |
W |
Sseg |
W |
Sseg |
W |
||
|
b -CdUO4 [central atom U(l): x = 0.000, y = 0.000, z = 0.000] |
||||||||||||
|
O(1) |
0.000 |
-0.278 |
0.000 |
1.90 |
17.7 |
18.8 |
17.9 |
18.8 |
18.3 |
18.6 |
15.8 |
15.9 |
|
O(1) |
0.000 |
0.278 |
0.000 |
1.90 |
17.7 |
18.8 |
17.9 |
18.8 |
18.3 |
18.6 |
15.8 |
15.9 |
|
O(2) |
-0.159 |
0.000 |
-0.500 |
2.08 |
15.7 |
15.3 |
15.2 |
15.1 |
12.8 |
13.6 |
8.8 |
9.5 |
|
O(2) |
0.159 |
0.000 |
-0.500 |
2.08 |
15.7 |
15.3 |
15.2 |
15.1 |
12.8 |
13.6 |
8.8 |
9.5 |
|
O(2) |
-0.159 |
0.000 |
0.500 |
2.08 |
15.7 |
15.3 |
15.2 |
15.1 |
12.8 |
13.6 |
8.8 |
9.5 |
|
O(2) |
0.159 |
0.000 |
0.500 |
2.08 |
15.7 |
15.3 |
15.2 |
15.1 |
12.8 |
13.6 |
8.8 |
9.5 |
|
U(l) |
0.000 |
0.000 |
-1.000 |
3.51 |
3.1 |
3.5 |
||||||
|
U(l) |
0.000 |
0.000 |
1.000 |
3.51 |
3.1 |
3.5 |
||||||
|
O(1) |
-0.500 |
-0.222 |
0.000 |
3.83 |
0.4 |
0.3 |
0.3 |
0.2 |
||||
|
O(1) |
0.500 |
-0.222 |
0.000 |
3.83 |
0.4 |
0.3 |
0.3 |
0.2 |
||||
|
O(1) |
0.500 |
0.222 |
0.000 |
3.83 |
0.4 |
0.3 |
0.3 |
0.2 |
||||
|
O(1) |
-0.500 |
0.222 |
0.000 |
3.83 |
0.4 |
0.3 |
0.3 |
0.2 |
||||
|
Cd(l) |
0.000 |
-0.500 |
-0.500 |
3.85 |
<0.1 |
<0.1 |
1.5 |
1.4 |
||||
|
Cd(l) |
0.000 |
0.500 |
-0.500 |
3.85 |
<0.1 |
<0.1 |
1.5 |
1.4 |
||||
|
Cd(l) |
0.000 |
-0.500 |
0.500 |
3.85 |
<0.1 |
<0.1 |
1.5 |
1.4 |
||||
|
Cd(l) |
0.000 |
0.500 |
0.500 |
3.85 |
<0.1 |
<0.1 |
1.5 |
1.4 |
||||
|
Cd(l) |
-0.500 |
0.000 |
-0.500 |
3.93 |
0.6 |
0.3 |
3.0 |
2.1 |
4.9 |
4.1 |
||
|
Cd(l) |
0.500 |
0.000 |
-0.500 |
3.93 |
0.6 |
0.3 |
3.0 |
2.1 |
4.9 |
4.1 |
||
|
Cd(l) |
-0.500 |
0.000 |
0.500 |
3.93 |
0.6 |
0.3 |
3.0 |
2.1 |
4.9 |
4.1 |
||
|
Cd(l) |
0.500 |
0.000 |
0.500 |
3.93 |
0.6 |
0.3 |
3.0 |
2.1 |
4.9 |
4.1 |
||
|
U(l) |
-0.500 |
-0.500 |
0.000 |
4.91 |
<0.1 |
<0.1 |
0.3 |
0.2 |
||||
|
U(l) |
0.500 |
-0.500 |
0.000 |
4.91 |
<0.1 |
<0.1 |
0.3 |
0.2 |
||||
|
U(l) |
0.500 |
0.500 |
0.000 |
4.91 |
<0.1 |
<0.1 |
0.3 |
0.2 |
||||
|
U(l) |
-0.500 |
0.500 |
0.000 |
4.91 |
<0.1 |
<0.1 |
0.3 |
0.2 |
||||
|
UO2(C4H4O5) [central atom U(l): x = 0.604, y = 0.396, z = 0.50] |
||||||||||||
|
O(4) |
0.524 |
0.326 |
0.608 |
1.79 |
19.9 |
21.5 |
19.9 |
21.3 |
19.2 |
20.2 |
17.9 |
18.3 |
|
O(4) |
0.674 |
0.476 |
0.892 |
1.79 |
19.9 |
21.5 |
19.9 |
21.3 |
19.2 |
20.2 |
17.9 |
18.3 |
|
O(2) |
0.663 |
0.648 |
0.653 |
2.37 |
11.4 |
11.4 |
11.5 |
11.3 |
8.1 |
7.5 |
4.3 |
3.2 |
|
O(2) |
0.352 |
0.337 |
0.847 |
2.37 |
11.4 |
11.4 |
11.5 |
11.3 |
8.1 |
7.5 |
4.3 |
3.2 |
|
O(1) |
0.888 |
0.385 |
0.683 |
2.43 |
13.4 |
12.2 |
10.7 |
10.2 |
6.3 |
5.6 |
3.1 |
2.2 |
|
O(1) |
0.615 |
0.112 |
0.817 |
2.43 |
13.4 |
12.2 |
10.7 |
10.2 |
6.3 |
5.6 |
3.1 |
2.2 |
|
O(3) |
0.383 |
0.617 |
0.750 |
2.56 |
8.5 |
8.2 |
8.5 |
8.1 |
2.3 |
2.3 |
||
|
C(l) |
0.935 |
0.267 |
0.622 |
3.22 |
3.4 |
2.9 |
8.2 |
8.4 |
12.0 |
12.6 |
||
|
C(l) |
0.733 |
0.065 |
0.878 |
3.22 |
3.4 |
2.9 |
8.2 |
8.4 |
12.0 |
12.6 |
||
|
O(2) |
0.837 |
0.148 |
0.597 |
3.25 |
1.1 |
0.8 |
0.1 |
0.1 |
||||
|
O(2) |
0.852 |
0.163 |
0.903 |
3.25 |
1.1 |
0.8 |
0.1 |
0.1 |
||||
|
C(l) |
0.565 |
0.767 |
0.628 |
3.33 |
0.1 |
0.1 |
4.1 |
4.3 |
8.0 |
8.8 |
||
|
C(l) |
0.233 |
0.435 |
0.872 |
3.33 |
0.1 |
0.1 |
4.1 |
4.3 |
8.0 |
8.8 |
||
|
C(2) |
0.233 |
0.597 |
0.815 |
3.52 |
2,8 |
2.8 |
4.0 |
4.3 |
||||
|
C(2) |
0.403 |
0.767 |
0.685 |
3.52 |
2.8 |
2.8 |
4.0 |
4.3 |
||||
|
C(l) |
0.767 |
0.565 |
0.372 |
4.60 |
<0.1 |
<0.1 |
0.7 |
0.6 |
||||
|
C(l) |
0.435 |
0.233 |
1.128 |
4.60 |
<0.1 |
<0.1 |
0.7 |
0.6 |
||||
* See footnote in Table 2.
angles, not exceeding 2.5% of the total solid angle of the uranium atom (4
p steradian), also correspond to these faces. Recall that a certain solid angle (W ), which is numerically equal to the area of a segment of a sphere of unit radius (with the center at the uranium atom) cut by the pyramid with the uranium atom in the apex and the corresponding face of the polyhedron in the base, corresponds to each face of the VDP of the A atom [4]. We believe that the solid angles are more important geometric characteristics of dual CPs, because unlike VS and Ss which change their absolute values with the variation of Kd(A-X), the solid angles of the faces are invariant with respect to the transformation of similarity as long as the combinatorially topological structure of the polyhedron does not change with the variation of Kd. In totality, the data available make it possible to suggest that all of the U-O contacts with W < 0.2p steradian [r(U-O) > 3 µ] correspond to additional non-valent long-range action, which was not found in the traditional method of the determination of atomic coordination numbers. The supplementary analysis shows that analogous abnormal contacts with r(U-O) > 3.0 µ and W (U-O)< 0.2p steradian occur in the dual CPs of five uranium atoms. Although the values of coordination numbers found for these dual CPs (7 and 8 for one and four compounds, respectively) coincide with well-known values, they must be considered as 6+1 and 7 + 1 from the classical point of view.Note in this connection that the experimental values of r(U-O) and
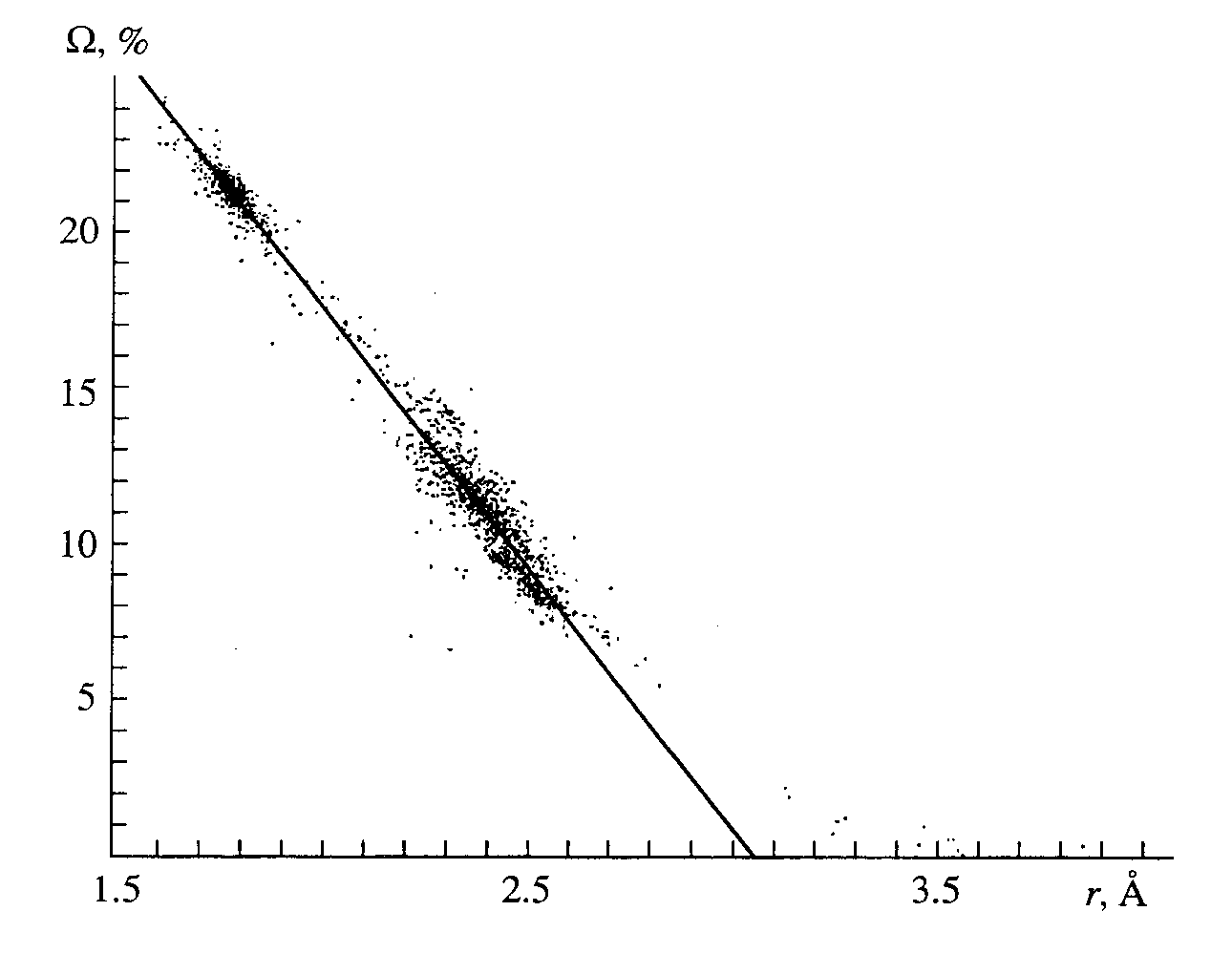
W (U-O) corresponding to all of the abnormal interactions at r(U-O) > 3 µ fall in the right nonlinear portion of the curve that describes the dependence of W (U-O) on r(U-O) in the VDPs of 354 uranium atoms in the structures of the compounds under discussion (Fig. 1). Contrary to the abnormally large distances, the values of r(U-O), which are considered as strong chemical bonds (covalent or polarized cova-lent) in the classical description of the structure of U(VI) compounds, lie in a straight line (Fig. 1), which may be expressed using the least-squares method asW
(U-O) = 51.2(1)- 16.79(5)r(U-O) (1)with the coefficient of correlation 0.991 for 2549 experimental points at r(U-O) < 3 µ. In equation (1) and in Fig. 1, the values of
W (U-O) are expressed as percents of the total solid angle (4p steradian) of the uranium atom, and r(U-O) are given in µ. The linear form of the function W (r) may be explained using the following approximations: (a) For the CP AXn, all of the distances from the A atom to the vertices of the dual CP are equal, that is, the dual CP is inscribed in the sphere of radius R with the center at the atom A. (b) The radii of the circumscribed spheres R are equal for all types of dual CPs, regardless of the coordination number of the A atom. (c) Each pyramid with the A atom at the apex that is based on the face of the dual CP corresponding to the A-X contact is approximated by a cone that has the area of its base equal to the area of the pyramid's face, and the length of its generator is R.
Fig. 1. Relationship between the solid angles of faces and r(U-O) in 354 VDPs of the U(VI) atoms.
In this case, it is not difficult to show that
W
(A-X) = 50 – 50 rf /R (2)where rf is the distance from the A atom to the face of the dual CP (the height of the cone). At Kd = 0.5 and rf= r(A-X)/2, formula (2) takes the form
W
(A-X) = 50 – 25 r(A-X)/R. (3)For the sampling considered, the value of R averaged over all of the VDPs of 354 uranium atoms is 1.72(4) µ; that is, the above-mentioned approximations (a) and (b) may be considered true. Some deviations in the values of coefficients in equations (1) and (3) are determined by the fact that the approximation (c) is rather rough. We believe that a small scatter in the values of R(
s = 0.04 µ) for the VDPs of uranium atoms suggests that the model considering the crystal structure of the uranium-containing compounds as a packing of deformable spheres [12] is correct. We emphasize especially that the plot shown in Fig. 1 is fully reproduced at any value of Kd(U-O) within the range 0 < Kd(U-O) � 0.5 because of similarity between the dual CPs of uranium atoms.According to the data obtained (Table 4), in the dual CPs of uranium atoms at Kd(U-O)
� 0.6, supplementary faces generally appear that correspond to nonvalent contacts U-X (X are atoms of the second coordination sphere) in addition to the faces corresponding to U-O bonds. The nature and total number of X atoms at fixed Kd(U-O) � 0.6 depend on the chemical composition and crystal structure of the compound. For a particular compound (some examples are given in Tables 2 and 3), the number of faces of the U-X type increases (generally to 18) as Kd(U-O) increases from 0.5 to 0.8.Table 4. Characteristics of the dual coordination polyhedra of U(VI) atoms at Kd(U-O) � 0.6*
|
Compo- Sition |
Kd=0.6 |
Kd=0.6 |
Kd=0.6 |
|||||||||
|
m |
N |
VS, µ3 |
SS, µ2 |
m |
N |
VS, µ3 |
SS, µ2 |
m |
N |
VS, µ3 |
SS, µ2 |
|
|
UO4Xm |
6,14 |
2 |
28.4(1.5) |
52.8(3.4) |
||||||||
|
UO5Xm |
7,12 |
3 |
27.5(2.4) |
51.9(2.8) |
||||||||
|
UO6Xm |
0-5,8 |
28 |
15.8(5) |
37.9(9) |
0-2,4-6,8, 10-12,14 |
31 |
24.5(1.0) |
49.3(2.3) |
0,4-6,8, 10-12, 14,16,18 |
41 |
32.5(3.0) |
57.7(4.5) |
|
UO7Xm |
0-8 |
212 |
15.7(4) |
36.8(7) |
0-5,7-11 |
212 |
24.3(9) |
48.6(1.4) |
1 -14,16 |
208 |
33.2(2.3) |
58.9(3.0) |
|
UO8Xm |
0,2 - 6, 8,9 |
108 |
15.7(4) |
36.3(7) |
2-11,14 |
109 |
22.9(1.0) |
46.7(1.5) |
2-14, 16,18 |
100 |
29.7(2.1) |
55.8(2.7) |
|
UO9Xm |
4 |
1 |
16.4 |
37.5 |
6 |
1 |
25.9 |
50.3 |
||||
|
UO10Xm |
0,2-4 |
4 |
16.3(5) |
38.5(5) |
0 |
1 |
26.1 |
53.2 |
||||
|
UO12Xm |
0 |
1 |
15.9 |
37.8 |
||||||||
|
UOnXm** |
354 |
15.7(4) |
36.8(8) |
354 |
23.9(1.1) |
48.1(1.8) |
354 |
32.1(2.8) |
57.8(3.5) |
|||
Notes:
* m are stoichiometric coefficients indicating the number of X atoms of the second coordination sphere contained in the dual CP UOnXm; N is the number of crystallographic sorts of U atoms with the value of n given in the first column.
** This line contains the average values of VS and SS, for all 354 sorts of uranium atoms regardless of the composition of CP UOnXm.
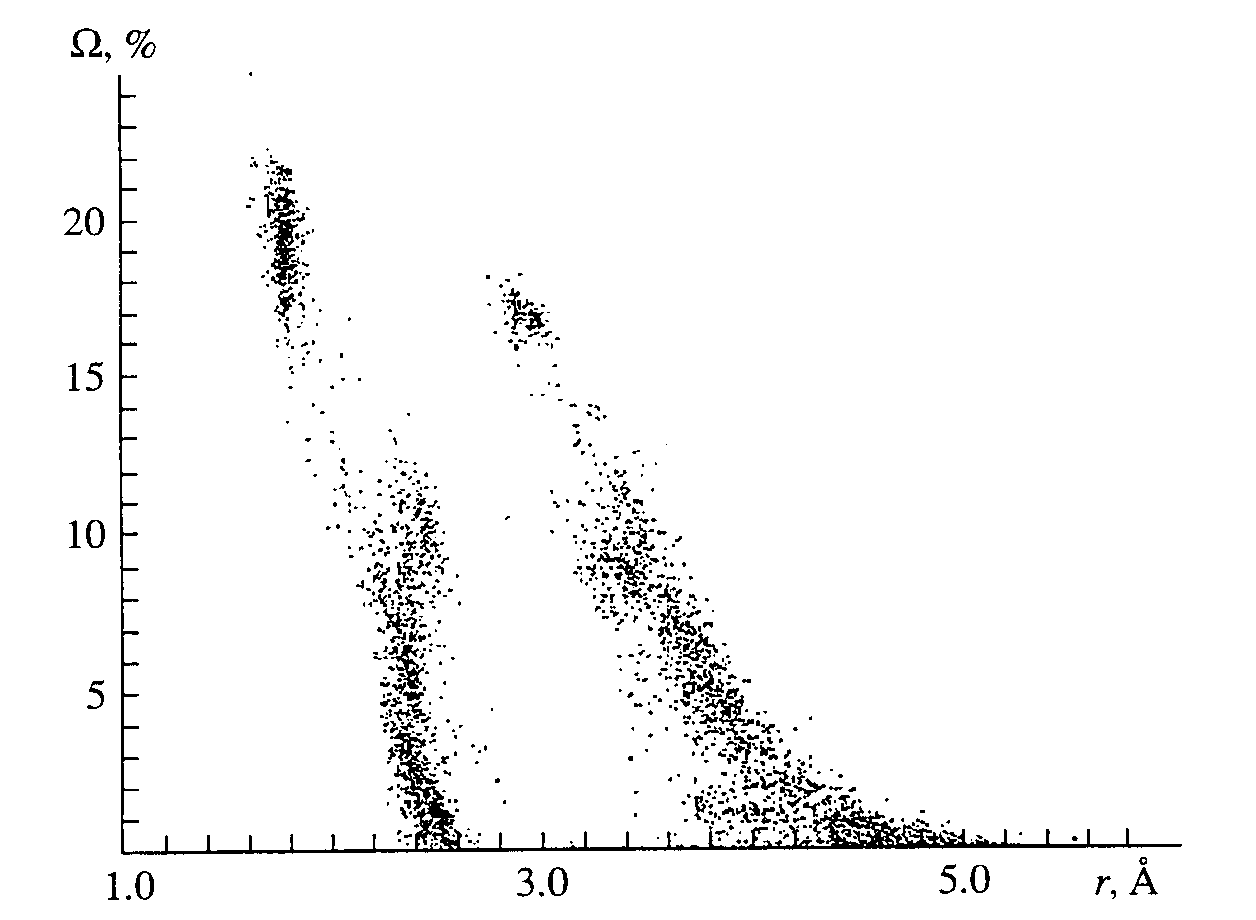
Fig. 2. Relationship between the solid angles corresponding to the faces of 354 dual CPs in UOnXm and r[U-O(X)] at Kd= 0.8.
The solid angles
W (U-X) of the faces that appear increase with increasing Kd(U-O) as a result of both a decrease in the solid angles of the faces of the U-O type (Table 2) and the disappearance of some faces of such type, starting with those that correspond to W (U-O) < 0.2p steradian (Table 3) even at Kd(U-O) � 0.5. As may be seen from Fig. 2 or from examples in Tables 2 and 3, the faces with the values of W (U-O) achieving 15 - 20% of the total solid angle of 4p steradian correspond to covalently bonded oxygen atoms of the first coordination sphere in the dual CP of composition UOnXm (4 � n � 8 and 0 � m � 18) even at Kd(U-O) = 0.8. Further analysis shows that, in spite of the wide variety of the composition (Table 4) and form of the dual CP of compounds UOnXm, rather compact regions of values correspond to X atoms of a particular chemical sort in the plot of the relationship between W (U-X) and r(U-X). As an example. Fig. 3 displays the fragment of Fig. 2 showing the relationship between W (U-X) and r(U-X) only for X = O or S at Kd(U-O) = 0.8 in all of the sulfurcontaining uranium compounds (32 out of 276 compounds) used in the calculations.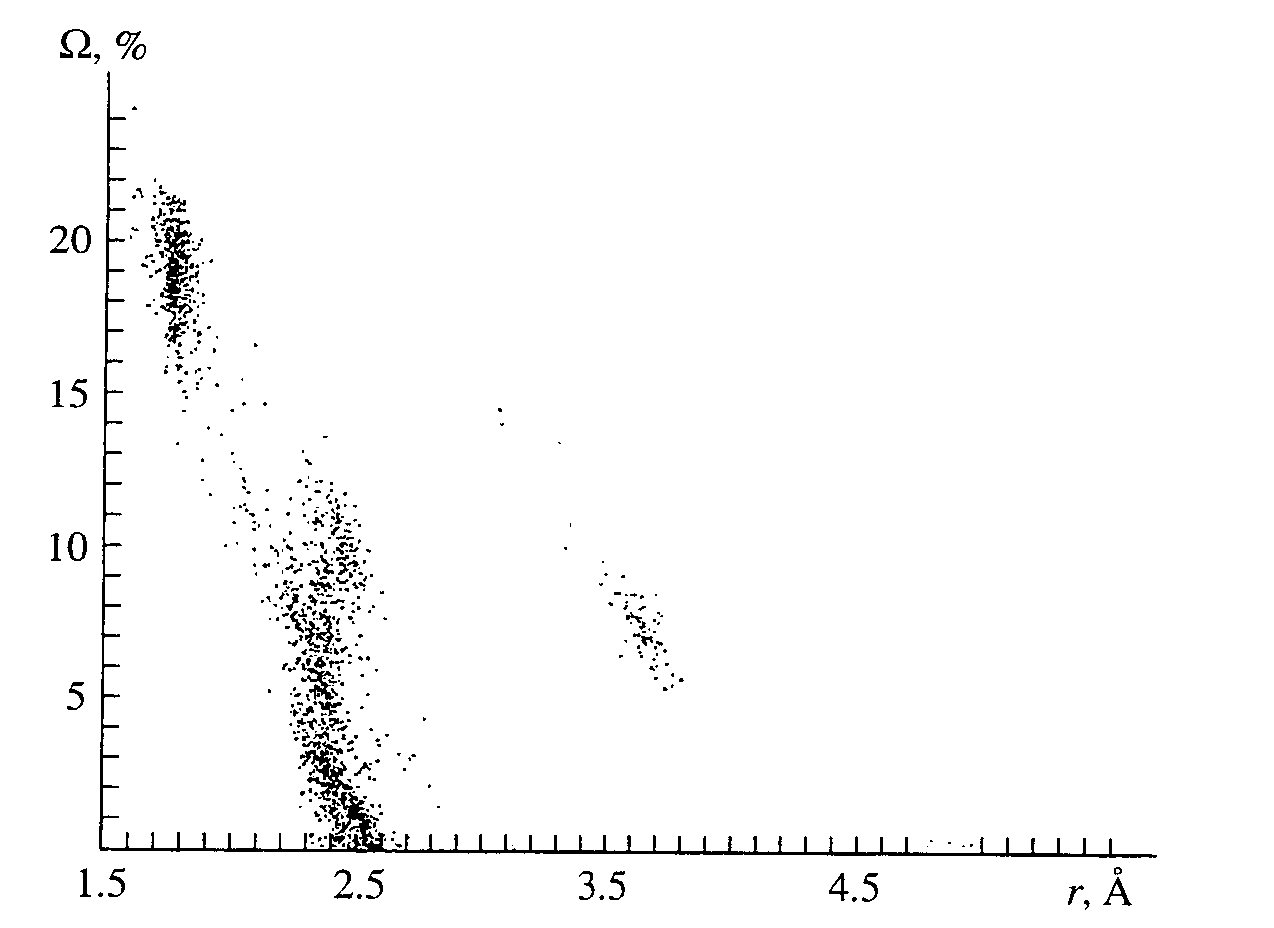
Fig. 3. Relationship between the solid angles corresponding to the faces of 99 dual CPs in UOnXm and r[U-O(S)] at Kd = 0.8.
The results presented above (Table 4) make it possible to suggest that all of the faces of the A-X type of the dual CP of a certain atom A at
W (A-X) > 0.2p steradian correspond to valence contacts A-X (that is, to strong chemical bonds) in the structure of any compound at Kd(A-X) � 0.5. If this suggestion, which possibly requires a refinement of the threshold value of W (A-X), is confirmed for other inorganic and coordination compounds, the direct calculation of the geometric characteristics of the dual CP at Kd = 0.5 for any bonds will make it possible to determine the classical value of the coordination number of any A atom in the structure without using any information about the radii of atoms in the compound.In the calculation of characteristics of the dual CP, the choice of constant value Kd = 0.5 for all of the bonds is determined by the following reasons. First, only at Kd = 0.5 are dual CPs coincident with VDPs, which are capable of filling three-dimensional space without hollows; at any Kd
� 0.5, the partition of the space by a procedure analogous to the construction of VDPs generally gives rise to simply connected spatial regions of points, which do not belong to any of the dual CPs. Second, for the heteronuclear bonds A-X, there is no criterion for choosing a "correct" Kd for a given pair of atoms. The use of any crystalline atomic radii for this purpose leaves the problem unsolved, because, as noted above, the concept of a radius itself is, to a great extent, a matter of convention, and it is not clear a priori which system of radii should be used for a particular pair of A and X atoms in the structure of a specific compound. Third, any value of Kd � 0.5 taken as a "correct" one for a particular A-X bond is strictly correct only at a single point Z corresponding to the intersection of this bond with the plane normal to it, because the value of Kd equals the ratio of the lengths of the AZ and AX segments. For any other point of this plane (for example, P), the AP : PX ratio will differ from the predetermined value AZ/ZX = Kd( 1 – Kd) and will depend on the length of the ZP segment. At the same time, the solid angles of the faces of any dual CP calculated at 0 < Kd < 0.5 coincide because of similarity to those calculated for the VDP of the same atom, i.e., at Kd= 0.5.The data obtained (Tables 1 and 4) show that, at any fixed value of Kd(U-O), the volumes of the dual CPs (VS) of U(VI) atoms are equal within the limits of twice the mean square deviation and are independent of the uranium coordination number. Thus, the mean volume of the VDP (VVDP), that is, VS at Kd = 0.5, of a uranium(VI) atom in the polyhedra UOn for 354 crystallographically different sorts of atoms equals 9.18(25) µ3. If only the dual CPs in which the number of oxygen atoms coincides with the classical coordination number of U(VI) (i.e., only the dual CPs of UOn or UOnXm types with n = 6, 7, or 8, which are appropriate to no less than 347 of 354 uranium atoms at any Kd
� 0.8) are taken into account, the corresponding values of VS overlap even within 1s (VS). This fact is especially remarkable because the volumes of the corresponding classical CPs (Table 1) increase significantly as the coordination numbers of uranium atoms increase. At any given value of Kd(U-O), the total area of the faces of the dual CP and VDP (SS and SVDP), the mean value of which equals 25.8(5) µ2 for 354 VDPs, is found to be constant (within 2s, Tables 1 and 4), as are VS and VVDP of the uranium(VI) atom. Note that the values of VS and VVDP (or SS and SVDP) given in Tables 1 and 4, in accordance with [4], are not the absolute characteristics of the regions of action of uranium atoms and are important only for comparison analysis, because the ratio of the volumes or surface areas of any two bodies are invariant with respect to similarity transformation.Note in conclusion that the results obtained corroborate the suggestion [4] of similarity between the VDP and the region of action of the U(VI) atom in the structure of a particular compound. The established fact that VVDP and SVDP are independent of the coordination number and the shape of the CPs of uranium atoms gives grounds to expect that U(VI) atoms in crystal structures (of oxygen-containing compounds at least) should be modeled not by rigid spheres but by soft spheres, capable of deforming under the action of atoms of the first coordination sphere and preserving, in this case, both the volume and area of the surface. In our opinion, it would be of interest to verify the above-mentioned conclusions using compounds bearing different complexing atoms in various oxidation states as an example. However, the systematic examination of the geometric characteristics of the VDPs of these atoms in structurally investigated compounds is required for this purpose.
ACKNOWLEDGMENTS
The authors are thankful to A.I. Yanovskii for the presentation of structural information from the Cambridge Databank.
REFERENCES