 MANUAL MANUAL
MANUAL MANUAL
 PLATON HOMEPAGE
PLATON HOMEPAGE
Coordinate input data can be either Cartesian or fractional, originating from crystal structure determination programs such as SHELXS, refinement programs such as SHELXL or otherwise.
PLUTON may be considered as a completely redesigned and considerably expanded decendent of the popular PLUTO(78) program by Motherwell and Clegg.
Drawings are produced either on a display surface (graphics terminal or workstation) called DISPLAY or as a meta-file saving graphics instructions (called META) for later use such inclusion in word processors (e.g. Word) or laser printer hardcopy, suitable for publication.
PLUTON is currently available only as a build-in link into the PLATON suite of programs.
A drawing of a molecular structure as generated by PLUTON may be thought of in three principle aspects: the content, the style and the viewpoint.
By default, connected and possibly symmetry expanded sets of atoms constituting molecules or ions will be assembled.
The atom types may be distinguished with various patterns in the black-and-white mode or colored to differentiate between the atom types.
Drawings may be made in parallel projection, perspective, or stereo-pair perspective. By default, a STICK-style drawing will be generated.
All input to the program is free-format, using (sub)keywords and numerical data. The input to PLUTON is normally provided in two parts: a disk file, in general produced by a program or prepared with a text editor and containing the pertinent data on the structure at hand such as atomic coordinates, followed by interactive input of the instructions via the keyboard or mouse-clicks.
In general, a small set of instructions in terms of global keywords with associated default parameter settings will suffice to produce a drawing for the set of supplied atomic coordinates. Alternatively, all input may be given on the disk file or interactively from the keyboard.
More than one data set, separated by an ENDS card may be present on the disk file. This is a convenient feature for browsing the structures retrieved from the Cambridge Structural Database. In that case, an interactively supplied END statement will not stop program execution but load the next data set (use the instruction STOP for premature abortion). Facilities are available (the SAVE instruction) to run the same set of instructions on a file that contains more than one data set (e.g. a series of structures extracted from the Cambridge Structural Database).
Note: PLUTON is not a finished program. It is extended and improved constantly on the basis of the needs that arise as part of our research work, including many hundreds of crystal structure determinations (organic, organometallic and inorganic) carried out as part of a national crystallographic service. New features are planned. Therefore there may be some discrepancies between the actual program and this manual.
Suggestions for improvement or information on errors are welcome.
A Tektronix emulator or the X-Window menu-off mode is assumed here. X-Window based implementations support both keyboard instructions and menu driven input with ain automatic redraw of an updated image.
The structural parameters are assumed to reside on a disk file, named cyto.spf in this example, for which the contents are listed below (free format). This particular input format is convenient for viewing structures with parameters taken from a publication:
TITL CYTOSINE (ANHYDROUS) ACTA CRYST. B29, 1234, 1973. CELL 13.044 9.495 3.814 90 90 90 SPGR P212121 N1 0.0222 0.0285 0.4410 C2 -.0164 0.1561 0.3276 O2 -.0998 0.1595 0.1710 N3 0.0402 0.2745 0.3877 C4 0.1308 0.2635 0.5492 N4 0.1842 0.3828 0.5967 C5 0.1705 0.1337 0.6674 C6 0.1134 0.0187 0.6084 H1 -.013 -.057 0.363 H3 0.154 0.463 0.541 H4 0.248 0.378 0.652 H5 0.237 0.128 0.796 H6 0.132 -.078 0.692PLUTON may be invoked for this data set with the command:
pluton cyto.spf
As a result the data set CYTO is loaded and, since this file does not contain an END instruction at the end of the file, the program comes, after that the end-of-file has been reached on this file, with the prompt >>, ready to receive additional data and/or instructions interactively.
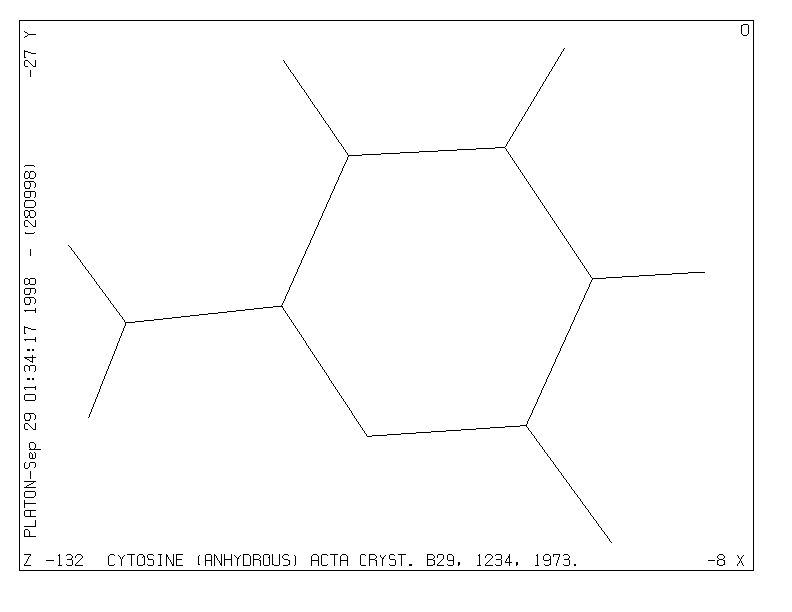
When we now type in respons to the prompt the instruction PLOT, the program will automatically assemble a unique molecule (i.e. symmetry operations will be applied when necessary to find connected sets of atoms), find all bonds and generate a minimum overlap stick-style plot of the structure (Fig. 1), optionally followed by a bell signal on completion of the plot. The program now waits for the user to press the RETURN (or ENTER) key to present the prompt >> again and to accept new instructions. Alternatively, a new instruction may be typed directly without waiting for the prompt.
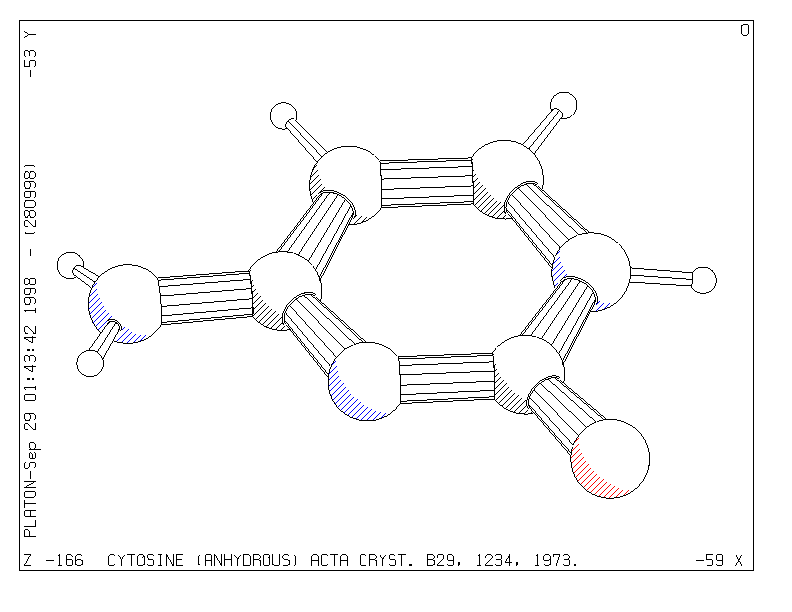
At this point the automatic minimum overlap view direction may be modified with any of the available VIEW instructions. As an example we can rotate the structure clockwise by 45 degrees about the horizontal X-axis with the instruction VIEW CURRENT XROT 45 (or to the same effect XROT 45) and see the effect with a new PLOT instruction. The style of the plot may be changed into the SOLID, STRAW, ROD or CPK style with corresponding instructions: e.g. ROD SHADE COLOR followed by PLOT ( Fig. 2) or CPK GLOBE COLOR followed by PLOT (Fig. 3).
A PLUTON session thus involves a series of cycles, each of which consisting of contents, style and viewpoint modification instructions, if any, followed by a PLOT instruction. As an aid, it is possible to inspect the contents of the various internal tables with instructions such as LIST ATOMS, LIST BONDS or LIST ARU. On- line information on the available instructions is available by typing HELP. A status line reports the current values on some of the more relevant settings. Use HELP GRAPHICS for instructions to change the default graphics settings.
The input set of fractional coordinates is not required to form a connected set or even a complete molecule in case of molecules sitting on a crystallographic symmetry element. Unless instructed explicitly by the user, the necessarily calculations are carried out automatically as communicated to the user by the expression AEX:: JOIN RADII UNIQUE EXPAND. This will automatically generate a connected set based on standard covalent radii and symmetry expanded when applicable. Where appropriate the user is informed about problems, new parameter values and the current status of relevant parameters.
Intermolecular hydrogen bonds of the type Donor-H...Acceptor may now be generated with the instruction:
JOIN HBONDS
An alternative would be
JOIN RADII INTER H 1.2 O 1.5 N 1.5
but this will generate many unwanted additional contacts that will have to be deleted subsequently from the bond list). The list options (e.g. LIST ATOM, LIST BONDS or LIST ARU) may be used following this instruction to verify that several items were added to the atom, bond and aru lists.
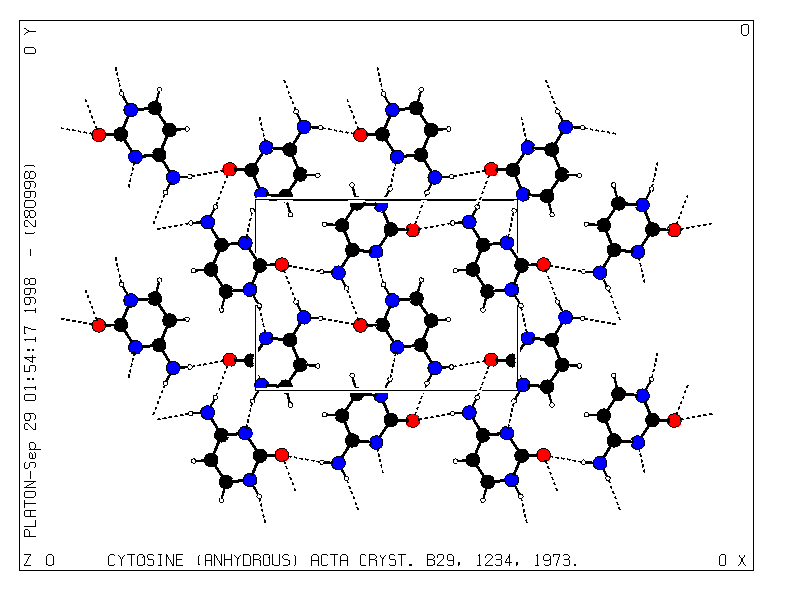
A SOLID type packing diagram with the unit cell outline and viewed down the c-axis of the structure is generated with a few additional instructions (Fig. 4):
SOLID
PACK RANGE -0.5 1.5 -0.5 1.5 0 1
UNITCELL
VIEW ZO
PLOT
To finish the program the instruction END should be issued. The
trailer listing file provides a detailed log of the current
session. All instructions are saved on a journal file to
reconstruct previous style and orientation settings. In addition
the name of the generated meta file (if any) is given.
The input atomic coordinates (x, y, z) are with reference to user- defined axes (a, b, c), which will usually be either crystallographic unit cell axes or an arbitrary orthogonal set; these coordinates are input as fractions of the unit cell edges or as Angstrom units (in the latter case they are converted to and stored internally as fractions of dummy cell edges). A second, orthogonal system (A, B, C) (see J.D. Dunitz, X-Ray analysis and the Structure of Organic Molecules, Cornell University Press, p235) with coordinates (XO,YO,ZO) in Angstroms is set up internally: The unit vector A is choosen along a, B as unit vector normal to a in the ab plane, and C normal to A and B. This orthogonal system is coincident with the first system when the input axes are orthogonal. The third system is the plotting coordinate system in cm: XP across the picture from left to right, YP up the picture from bottom to top and ZP out of the paper. All these axial sets are right-handed and absolute configuration is preserved in all rotations.
As atoms are input to the program, they are stored both in the X, Y, Z and the XO, YO, ZO coordinate system. The XP, YP, ZP system is used only when plotting is actually being performed. Each atom has additional information stored for it including the atom name (the embedded element name is used by default to set various radii) and various flags such as for inclusion in the plot and labelling. This data structure is known as the atom list. Duplicate entries with the same coordinates are skipped from the input stream.
The JOIN instructions (either by default or as specified) set up a list of connections (known as bond list) between atoms. Each connection is simply a pair of atom serial numbers referring to the input atom list. Some JOIN instructions also find connections between input atoms and other atoms related to the original list by crystallographic symmetry. This may include both connections to a symmetry related part of a molecule sitting on a special position or intermolecular connections. In order to store these connections for plotting, the program appends dummy symmetry-generated atoms to the atom list and marks them with a special flag.
If the final picture is to contain several molecules, as in a packing diagram, or a complete molecule for which the input atoms represent only the symmetry-independent portion of it when a molecule lies in a crystallographically special position in the unit cell, the program will have to generate XP, YP, ZP coordinates for the atoms in each of these molecules or symmetry-related molecular fragments. This is controlled by two further lists: a list of symmetry operations pertaining to the structure, input directly using the SPGR or LATT and SYMM instructions; and a list of 'molecules and ions' (known as ARU list) to be drawn (see discussion on ARU & ASU below).
The program works as follows. Instructions (free format) are read and interpreted. Various instructions add to the list of atoms and symmetry operators. The JOIN instructions set up the connections list and may also add to the atom and ARU-lists if connections between ARU's are to be generated. Plotting parameters are then set by user instructions or left at default settings. No plotting is actually done until the PLOT instruction is read. This first sets up the XP, YP, ZP plotting list of all atoms to be plotted. The information in the ARU list and the current view matrix together tell the program how to convert X, Y, Z into XP, YP, ZP. The plotting coordinates and atom radii are scaled from Angstroms to cm. and corrected for perspective if requested. In the non-stick mode, all atoms in the plotting list are drawn, allowing for obscuring of some by others and bonds, and intersection of spheres in space-filling models and bonds in the ROD mode, except that dummy atoms are not usually drawn - they are required only so that connections between molecules are possible, and will generally be duplicated by other non-dummy atoms generated in the XP, YP, ZP list by symmetry from the original input atoms. Following the atoms, all the bonds are drawn, omitting portions obscured by atoms or bonds. Atoms are labelled by a routine which minimises overlap of the labels by atoms, bonds and other labels. When the picture is complete, the next instruction may be read.
Most data are stacked in a large array, either bottom-up or top down, so that the stack size is the limiting factor for the size of the problem that the program can handle. The program reports on the maximum memory usage in the current run. The stack size is a program implementation parameter.
The input list of atoms is checked on redundancy.
The order in which data are read is as follows:
Disorder. Based on the supplied population parameters attemps are made to suppress bonds between disorder parts with unequal population parameter values.
By using the default settings associated with the choosen plot style (e.g. ROD) only a few of the available instruction types are absolutely required to produce a preliminary drawing of a structure. The absolute minimum input is a set of Angstrom coordinates, a PLOT instruction and END to stop the program.
In the description of individual instructions below the following applies:
The angle between the three specified atoms with atom-name2 as apex is calculated. The three atoms are not necessarily bonded atoms, but should be present in the current atom list. This list can be inspected with a LIST ATOMS instruction. An individual angle may be calculated as an aid in the analysis of the geometry of the structure. Exhaustive geometry listings may be obtained with the companion program PLATON. (See olso the GEOM instruction)
Example: ANGLE C(2) O2 N(2)
The following data are in orthogonal Angstrom units after multiplication with 'scale'.
The 'scale' should be adequate riIn order to fit in the automatic connectivity search based on the build-in covalent radii.
This instruction adds in a cumulative fashion asymmetric residue units (aru's) to the list of aru's to be drawn. It allows the user to have direct control over the list of residues to be drawn. This instruction is needed only when the automatic maintenance of this list with the JOIN RADII and/or PACK instructions is found to be not adequate. The ARU list may be inspected with the LIST ARU instruction. ARU's (e.g. aru1) are designated by a composite number (ARU) that may be split up into five parts: ARU = S*1000 + TX*100 + TY*10 + TZ + 555 + R*0.01, where S is the serial number of the symmetry operator (from the complete list to be inspected with a LIST SYMM instruction) to be applied to the input list of coordinates; TX, TY, TZ are the unit cell translations and R is the residue number. A negative sign of ARU means that the specified residue should not be drawn in the following plots, and may be used to eliminate residues temporarily from the list of residues to be plotted. This may useful in conjunction with the pack instruction.
Often residue numbers are not explicitly indicated and the fractional part of ARU effectively set to zero. In this form the complete asymmetric structure unit (ASU) is meant.
Examples: ARU 3555 7564 -1555 ARU 3564.01
Note: for packing reasons, the number of residues that can be coded is restricted to 9 with corresponding change in aru coding.
The effective ARU list is emptied with the NONE sub-keyword. (A LIST ARU will show that all ARU's now have negative values). The list is reset to what it was after a previous JOIN RADII UNIQUE or INTER instruction with the UNIQUE or INTER keywords respectively. A RESTORE sub-keyword resets all negative entries in the molecule list to their absolute value, so that they are active again.
If the effective ARU list is empty when the PLOT instruction is read, a ARU 1555 instruction will be executed automatically.
Example: ARU NONE
By default the drawing will be surrounded with a rectangular box outline. This setting may be changed with the ON and OFF sub- keywords. The structure is drawn with a shrinkage factor of 90 percent relative to the automatic scale factor that fits the drawing within the BOX-outline to provide a margin. This default percentage may be changed with the SHRINK shr option. This option may be also of use when a drawing which a non-default scale is required. The three numbers shown in the bottom right, top left and bottom left corner of the box are the cumulative rotation angles xr, yr and zr respectively. These numbers may be used to reconstruct this particular orientation in a later session directly from the default UNIT orientation via a VIEW UNIT XR xr YR yr ZR zr instruction. The default horizontal to vertical size ratio of the box is 4/3. A ratio of 1.0 results in a square box.
Example: BOX RATIO 2 SHRINK 80
An alternative to color coding of atom types is a distinction of atom types with different patterns drawn on the spheres representing the atoms. This is useful for black-and-white hardcopy and publication drawings. The current (default) settings may be inspected with the LIST TYPES instruction. Available options for bwc are contour, net, shade, segment, dots, black, cross, parallel, globe, meridian, horizontal, vertical, mesh, diagonal, slant, textile and void.
Example: BWC Pd GLOBE
BWC ON/OFF toggle.
This instruction may be used to change the default color (normally BLACK for plotters & laser printers and WHITE for terminal screens) of the plot into the one chosen (subject to the availability of colour with the actual graphics facility). This color is used for bonds, atom circumference and text.
Example: COLOR RED
The color setting of an atom-type, as shown with a LIST TYPES instruction, may be modified with the COLOR TYPE instruction. Available values for col are: BLACK(WHITE)/RED/GREEN/BLUE/YELLOW/ORANGE/VIOLET/BROWN.
Example: COLOR TYPE C black O red
On/off toggle for display of residue colors.
On/off toggle for color per ARU.
Color on/off toggle.
List geometry about 'atom-name'
This sets parameters for a space-filling model (Corey-Pauling- Koltum) with atomic radii that are taken by default from internal tables and optionally with RADII ATOMS instructions. The surface may be either globally Shaded, Coloured, Dotted, drawn with Contours, segments, a Net structure etc. or with a pattern dependent on the atom type (BWCOL). The keyword SHADE causes the drawing of shade lines representing shadow from a light source whose position is given by the two angles a1 and a2. The setting of both angles to zero models a light source coming directly towards the viewer along the ZP axis, so that the whole atom is shaded. The angles a1 and a2 are rotations of the light source about YP and ZP respectively and in that order, and d is the spacing in cm between shading lines. Default settings are 120, -45, 0.15.
The atoms may be coloured as a function of their atom type. This feature is implemented as the SHADE option with a1=a2=0 and d = 0.05 in the absence of other sub-keywords with the COLOR sub- keyword.
The NET sub-keyword produces a NET with two sets of perpendicular great-circles drawn on the surface (colored in combination with the COLOR sub-keyword). the number of horizontal and vertical circles may be modified with nh and nv.
The GLOBE sub-keyword produces a polar grid surface, combined with shading.
The CONTOUR sub-keyword produces a set of parallel circles on the surface.
The BWCOL sub-keyword may be used to differentiate between atom types in a black-and-white plot.
The SPOT sub-keyword asks for a light reflexion spot on the surface.
The STICK keyword produces a CPK plot combined with a STICK frame to show the chemistry of the molecule.
Example: CPK NET COLOR SPOT
Note: CPK COLOUR DOTS will generate white dots on the colored atomic spheres.
CROTX will produce a stick model rotating about the horizontal X- axis. CROTYM gives a model rotation about y, anti-clockwise). Bonds may be colored as a functions of the atomtypes at their end-points with the COLOR sub-keyword.
Example: CROTX COLOR
This instruction allows the elimination of bonds from the bond list, such as those resulting from intermolecular contact searches or between non-bonded metals.
Example: DETACH CA C
The deletion of undesired intermolecular bonds may require special treatment. The first step involves finding the exact entry in the bond-list (use LIST BONDS) to be deleted.
Example: to delete the bond O1 H1A a from the list, issue DETACH O1 H(1A)a.
Replace set of bonds by one bond to the centre of gravity of the specified atoms. This is particularly useful to represent the eta-5 bond of a metal to a cyclopentadienyl ring:
Example: DEFINE Ti TO C1 C2 C3 C4 C5
Delete specified items.
The distance between the specified and not necessarily bonded atoms is calculated. Both atoms should be in the current atom list (to be checked with a LIST ATOMS instruction) or explicitly generated using the optional aru designator.
Example: DIST C1 C3 DIST C1 C3 2555.01
End of plot instructions for this data set. The next data set following an ENDS card is loaded when the SPF-file contains multiple data sets. The program terminates when no data are left; information on the produced files will be shown. Direct termination is achieved with QUIT or STOP instructions.
Instruction to manage multi-entry data sets (e.g. from CSD).
Exclude the specified atoms from the atom list. ORIG means all the atoms originally input; UNIQUE means atoms generated by JOIN RADII UNIQUE; INTER means atoms generated by JOIN RADII INTER. Bonds are only drawn between included atoms, except that bonds to UNIQUE and INTER atoms are drawn even if these atoms are omitted. Usually, generated atoms need not be specifically included because they duplicate original atoms in generated molecules. The effect of successive INCLUDE and EXCLUDE instructions is cumulative. INCLUDE ALL and EXCLUDE NONE are synonymous; so are INCLUDE NONE and EXCLUDE ALL. The default is INCLUDE ALL.
Example: EXCLUDE H
Interactive calculation of geometry around atom, i.e bond distances and bond angles.
This gives on-line help on the specified type of instructions. In particular HELP GRAPHICS will inform on the way the graphics is implemented. HELP SPGR gives a listing of all the space group names known to the program. HELP ALL gives the full list of available instructions.
Example: HELP STYLE
Include the specified atoms in the atom list. See EXCLUDE.
Instruction modifies defaults to suitable values for inorganic compounds.
A PLOT instruction that, since the start of the program or after a RESET, was not preceded by any JOIN instruction will automatically invoke the execution of a JOIN RADII UNIQUE EXPAND instruction. This automatically produces a list of connections, an ARU-list and an atom list for the possibly symmetrical molecule(s) in the structure, based on internal covalent atomic radii. All distances between two, possibly symmetry transformed, atoms less than the sum of the covalent radii for the two atoms plus a tolerance (by default tol = 0.2 Angstrom per atom) will be entered in the connection list and related changes or additions are made to the molecule and atom lists. Atoms are moved (unless NOMOVE disables it to do so) to symmetry-equivalent positions in order to form connected fragments. If the molecule has symmetry coincident with space group symmetry operators and only the asymmetric coordinate set supplied, the program will look for connections between the symmetry-related portions of the molecule. This involves the generation of dummy atoms and modification of the molecule list as well so that the PLOT instruction will show the complete molecule. The radii used for the automatic JOIN instruction can be inspected with the LIST TYPES instruction. The user may override this automatic feature by explicitly specifying the required JOIN instruction(s) before the first PLOT instruction. When the EXPAND sub-keyword is left out the molecules will not be fully symmetry expanded as is needed for molecules exceeding threefold site symmetry. Symmetry is not taken into account when in addition to this the UNIQUE sub-keyword is left out so that the user is held responsible to provide the correct set of atomic coordinates assumed to be already in bonding distance. Also the atomic radii used may be changed by their explicit specification:
JOIN RADII C 0.85 BR 1.35 H 0.4
This will find all C-C bonds less than 1.7, C-H less than 1.25 and C-Br less than 2.2 Angstrom. Tolerance values are not added to explicitly specified radii.
The ARU list is reset to the input set before entering the connection search routine when a JOIN RADII instruction is read and the connection list is also emptied.
The TOL sub-keyword may be used to change the value of tol to be used along with the radii drawn from the internal tables (see Appendix V). The single keyword instruction JOIN is equivalent with the expanded form JOIN RADII UNIQUE EXPAND.
When appropriate an additional 0.7 Angstrom is added to the tolerance value in the automatic radii mode to catch (earth-) alkali to non-metal contacts.
To generate intermolecular connections (e.g. Hydrogen bonds), the keyword INTER must follow RADII. This also involves generating dummy atoms and modifying the molecule list so that a subsequent plot will show several molecules unless the list is changed by a MOLES or PACK instruction.
Example: JOIN RADII INTER N 1.5
would form the unique molecule first and then find all potential hydrogen bonding interactions between nitrogen atoms less than 3 Angstrom. Alternatively, the sub-keyword HBONDS may be used for which only H to acceptor contacts are generated:
JOIN RADII INTER HBONDS
For JOIN RADII INTER, the molecule list is reset to the list generated by a previous JOIN RADII UNIQUE; the connection list remains at the current setting and the intermolecular connections are added to the list as they are found.
When no explicit radii are given on a JOIN RADII INTER card the program will use radii equal to the covalent radii + 0.8 + tol. When only part of the inter radii is specified it is implied that the radii for the remaining atom types is to be set to zero. The instruction JOIN HBONDS is equivalent to the expanded form JOIN RADII INTER HBONDS.
The first atom on the JOIN card is joined to each of the others.
Example: JOIN Mn1 TO C1 C2 C3 C4
This instruction sets up connections explicitly, adding them to those already existing. Each atom is to be joined to the one preceding it and the one following it in the list. Thus to draw a benzene ring with atoms C1, C2, C3, C4, C5 and C6:
JOIN C1 C2 C3 C4 C5 C6 C1
Option for dashed bonds.
A JOIN INTRA instruction deletes intermolecular connections (those generated by JOIN RADII INTER), leaving only intramolecular bonds, including those between symmetry-related parts of a molecule (generated by JOIN RADII UNIQUE).
JOIN NONE empties the connection list.
The specified atoms are added to the current list of atoms to be labelled. The program tries to find a suitable position for the label close to the atom giving minimum overlap by other plot items. The sub-keyword MOLES causes the symmetry code of the molecules to be plotted against the first atom of each molecule.
Examples:
LABEL Cu N O
LABEL C1 C2 C3 N4 C5
LABEL ARU
LABEL ALL
UNLABEL *
The effect of successive LABEL instructions is cumulative.
These label instructions are global. The ALL sub-keyword gives labelling of all atoms and is undone with UNLABEL ALL. ON and OFF have the same effect. Atom labels may be plotted with or without (default) parentheses.
This causes some of the lists held by the program to be displayed for inspection. The same lists are sent automatically to the trailer output file when a PLOT instruction is executed.
The ATOMS option gives a list of the input atoms together with any dummy atoms generated by a JOIN instruction. For each atom, the coordinates and atom radius are shown, together with flags to show whether the atoms are currently included and/or to be labelled.
BONDS list all the (active) connections currently held with their distance and plot style parameters.
LINES lists the information set up by RADII BONDS instructions.
MATR gives the current orientation matrix and the three decomposition angles to reconstruct this orientation with a VIEW UNIT XROT xr YROT yr ZROT zr instruction.
ARU gives the list of molecules.
STATUS list various parameter settings and other relevant data.
SYMM gives a list of all equivalent positions, preceded by their internal sequence number that is used in conjunction with the ARU instruction.
TYPES gives a listing of all atom-type dependent setting such as radii and colours. If none of the above keywords is given, all the information is shown.
Example: LIST ATOMS BONDS
This lists atom data from the atom list for the specified atom or atom-type.
Example: LIST ATOM O
This lists bond data for the specified bond from the bond list.
Example: LIST BOND C3 C4
This displays the current cell parameters for inspection.
List symmetry operations and related data.
Option to list an internal parameter value held in the PAR list. A range is listed when two numbers have been specified or the full list is case that none was specified. New values may be assigned with the SET PAR instruction.
Example: LIST PAR 5
Option to list an internal parameter value held in the IPR list. A range is listed when two numbers have been specified, or the whole list is case of no range specification. New values may be assigned with the SET IPR instruction.
Example: LIST IPR 3 5
A single perspective drawing is to be produced as seen from a point at d cm. along the ZP axis, above the centre of the drawing. The default setting is 10000 cm. (effectively infinity) but may be set to 60 cm. for convenient perspective viewing. The actual value is plotted in the top right corner of the plot (zero for infinite perspective) to avoid confusion.
Nomove toggle.
No atom list sorting toggle.
All atoms with their centres outside the box defined by the fractional coordinates xmin,...,zmax are omitted from the drawing, together with any bonds to them. This is useful as a 'window' on a packing diagram. The window may be reset by another OMIT OUTSIDE instruction, or cancelled alltogether by OMIT OUTSIDE 0. Note that molecules lying across the edge of the window will be drawn incomplete. An alternative option is the exclusion of all atoms with their centres outside a sphere with radius r around a specified atom.
Example: OMIT OUTSIDE C(1) 5
OMIT OUTSIDE 0.5 1 0.5 1 -.25 .25
Omit the specified items from the 'plot-list'.
Set default parameters for type organic structure.
The overlap margin (non-bonded bond-bond, atom-atom, atom-bond overlap) is set by default to the value mrg = 0.1 cm and the incoming bond shadow margin to shad = 0 cm. Overlap calculations (by default) may be turned off for fast testruns. This should be specified before plotting and after the specification of the plot-style.
Example: OVERLAP OFF
This causes a search for all ARU's having their centres inside the box defined by the fractional coordinates given. By default PACK RANGE displays the contents of a unit cell. However, this will not show the complete packing arrangement of the crystal; it is necessary to supply a range that will ensure that a pair of translated molecules will be found in each axial direction. This is achieved by giving a range of cell translations. In most structures, the result of a range of two in each axis is usually too congested to be of any use, so that it is best to restrict the range to one on the axis closest to the view direction.
e.g. PACK RANGE -0.5 1.5 -.5 1.5 0 1
might be appropriate for a z-axis projection. The instruction PACK generates by default all ARU's with their centre within the unit cell.
The ARU's that obey the conditions are added to the ARU list. The ARU list is reset to ARU UNIQUE at the beginning of the search. No other lists are affected. The ARU list may be displayed by the LIST ARU instruction.
ARU's are put in the moles list that satisfy the condition of lying within the range xmin,...,zmax and in the neighborhood of the specified hkl-plane (d1 and d2 are in Angstrom).
Example: PACK PLAN 1 1 1 -.5 .5 RANGE -1 2 -1 2 -1 2
This produces a drawing using the information set up by the previous instructions on either the current graphics medium (default) or on the specified medium (Display, Plotter or Meta- file). The default medium is either the one previously set or the display by default. An automatic JOIN RADII UNIQUE EXPAND instruction is executed when no previous JOIN instruction was given and a VIEW MIN instruction is case of no previously given VIEW instruction. The MOGLI option may be used to generate DGE and OBY files to be used to view the molecule in 3D with the PSSHOW program of the MOGLI package on a PS300 display (use PLOT CAL MOGLI to generate the files without display or plotter output).
The special instruction PLOT CAL 3 will generate a file with full 3D vector information for a ROD-style plot, independent of previous parameter settings.
position: N, NE, E, SE, S, SW, W, NW, NUCL, AUTO
The automatic label positioning routine considers eight (or nine in the case of SOLID NUCL) possible positions for each label. These are conveniently designated by the compass directions N, NE, E, SE, S, SW, W, NW (or NU). The PUT instruction forces particular atom labels into positions chosen explicitly by the user. Atoms named here are positioned first, then the automatic routine places the rest. A position may also be given as AUTO, so that all atoms of a given type can be fixed and then specific exceptions made:
Example: PUT C SE C12 W C14 AUTO
PUT instructions must follow LABEL. They will be ignored if they precede it.
This gives immediate exit from the program.
This globally sets the atomic radii (Angstrom) for the plotting of the spheres in the SOLID, ROD and CPK style. The appropriate instruction is normally executed automatically. In the SOLID and ROD mode spheres are drawn at 0.5 times their covalent radius and in the CPK mode at the covalent radius + 0.8.
This instruction may be used to input user defined radii for the spheres to be drawn. The actual values may be listed with a LIST TYPES instruction.
Bonds in SOLID or ROD drawings have cylindrical radius r Angstrom (default 0.04 and 0.2 resp.) and are drawn with n (default n = 8) lines along the circumference of the cylinder, spaces (180/(n+1)) degrees apart on the visible side of the cylinder. This instruction resets r and n for all bonds.
This sets r and n for all bonds to a particular atom or atom type. This may be useful to emphasize coordination of a particular atom. To produce single-line bonds, set r - 0 and n = 1. Bonds with n = 0 are omitted.
This sets r and n for a particular bond already in the connection list.
This sets r and n for all intermolecular bonds created by a JOIN RADII INTER instruction. This is useful, for example, in distinguishing hydrogen bonds.
RADII BONDS ALL not only resets r and n for all bonds, but also cancels all other RADII BONDS instructions which have already been given, i.e. 'all' really means all. To reset r and n for all bonds EXCEPT special ones, use this instruction instead.
When the program is drawing the bonds, it looks though the list of the RADII BONDS instructions and assigns the r and n values of the last instruction given which is relevant to the bond being drawn. In this way the result of conflicting instructions can be controlled by the order in which the data card are given. An example of such a list of instructions:
RADII BONDS ALL 0.05 5
RADII BONDS INTER 0.02 2
RADII BONDS TO CU1 0 1
RADII BONDS TO NA 0.03 2
RADII BONDS CU1 O4 0.06 10
Note, however, that if a particular RADII BONDS instruction is given a second time with different values of r and n, this resets r and n without altering the order of the RADII BONDS instructions. E.g., the sequence
RADII BONDS TO H 0.02 4
RADII BONDS INTER 0 1
RADII BONDS TO H 0.03 7
is equivalent to
RADII BONDS TO H 0.03 7
RADII BONDS INTER 0 1
i.e. the second RADII BONDS TO H instruction directly replaces the first.
In order to enhance the perspective effect of drawings (see MONO and STEREO options), bonds are drawn with an exaggerated taper, the degree of exaggeration being set by the parameter taper. taper = 2 gives maximum exaggeration. The default setting is 0.250. Tapering is applied even if the viewing distance is effectively infinite, unless taper = 0.
Before any plotting instructions are read, various default parameters for plotting must be set. This is automatically done at the beginning of the program, but can also be done at any time by use of RESET.
Labels are retraced n times.
This prepares for a ROD-style plot with atoms optionally shaded, coloured, drawn with a net or contoured surface or individually characterized (see CPK for further information).
This instruction may be used to save instructions to be used on other data sets as well. This feature is useful when examining a series of structures taken from the crystallographic data base. All instructions following a SAVE instruction until an END are saved.
Plotstep and substep are parameters that determine the accuracy of the plot, the speed and the computing time needed for the plot. Testing for overlap is done in steps of size plotstep. When a problem is encountered the testing will be redone in steps of size plotstep/substep. The default values are 0.1 cm and 4 steps respectively. For accurate plots, in particular on either the plotter or the meta-file, values of 0.01 cm and 1 are recommended for the above parameters.
This instruction is not meant for general use. It provides a facility to modify internal parameters, in particular those with no equivalent (sub)keyword.
The default character size (ch = 0.25 cm) may be changed with a SIZE 0 CHAR ch instruction.
Similarly, the default character size of the title (ti = 0.4 cm) may be changed with the TITLE sub-keyword: SIZE 0 TITL ti.
The cm/Angstrom scale may be specified with SCALE
Example: SIZE 0 SCALE 1.0
This gives a vintage-PLUTO-style bond-and-stick plot.
See the CPK instruction for other keywords.
Example: SOLID COLOUR CONTOUR 0
for a colored atom outline. (nc = number of contours)
This produces a stereo pair, as seen by an observer with eye- separation of 6 cm. at a distance d ( default 60) cm along the ZP axis. If the keywords RG or GR are present, the left-eye and right- eye views are superimposed instead of being placed side-by-side, but are drawn in different colors, so that the stereo effect can be observed using appropriate red/green or green/red filters. The sub- keyword CROSSED allows for crossed eye vision.
This asks for a stick model drawing, i.e. one in which the atoms are points and the bonds single lines. A succession of such drawings requires very little computing and plotting time, and is useful as a preliminary run for checkout and to find a good view direction.
Note: since atoms are represented by points in this presentation, as are bonds when viewed along their direction, non-bonded atoms and some linear molecules viewed from the end are not shown in STICK plots.
This gives an immediate EXIT from the program.
The title of the plot is normally taken from the TITL card that preceded the coordinate data. It can be overruled with a new TITL instruction.
The dihedral angle (not necessarily involving bonded atoms) for the specified atoms is calculated as an aid to analyze the geometry of the structure (exhaustive geometry listings may be obtained with the companion program PLATON).
Example: TORSION C(2) O2 N(2) C(4)
This allows a unit cell outline to be drawn. The outlines are
treated as bonds (in terms of radius and number of lines
drawn on the bond).
Their default setting is: rbo = 0.01 and nli = 2.
This instruction may be used to unlabel. see LABEL.
This produces a minimum overlap view. The view is perpendicular to the least-squares plane through the included atom set as generated for the current ARU list. A VIEW MIN instruction is executed automatically with a PLOT instruction when no previous VIEW instruction was given.
Example: VIEW MIN XROT 45
This produces a view with XO along XP (horizontal) and YO along YP (vertical), possibly modified with a sequence of rotations.
A given orientation may be reconstructed, independent from its history, using the three rotation values that are shown in the lower right corner (xr), the upper left corner (yr) and the lower left corner (zr) with the instruction:
Example: VIEW UNIT XROT xr YROT yr ZROT zr
VIEW is equivalent with VIEW UNIT.
This instruction updates the current view matrix. VIEW rotations is equivalent with VIEW CURRENT rotations.
Example: VIEW CURRENT XROT 45 ZROT -60
Note: XROT 45 is a shortcut equivalent for VIEW CURRENT XROT 45
View along one of the orthogonal axes towards the origin.
For a view along the line from the point (1.0, 0.5, 0.5), (0.5, 1.0, 0.5) or (0.5, 0.5, 1.0) towards the origin (crystal coordinates).
This causes an automatic ZROT sufficient to bring the projection of the line between the two atoms parallel to the plotting XP (YP) axis. A warning message is issued when the two atoms are superimposed in the current view, so that this instruction is not uniquely defined.
Example: VIEW ALIGN C1 C2 WITH YP
Note: to bring the vector between the atoms atom_name1 and atom_name2 parallel to XP use the instruction VIEW LINE atom_name1 atom_name2 YROT 90
This defines a view along the line from the point (x,y,z) towards the origin (crystal coordinates). This may be useful for views down the crystal axes for non-orthogonal crystal systems.
The direction of view is from the first atom to the second.
The view is into the angle n1-n2-n3 with n2 deepest in the plot.
The view is perpendicular to the plane containing the three atoms, seen from the side which makes a clockwise order of the atoms in the plot n1-n2-n3.
An inverted image with respect to the current image is obtained with this instruction.
This option allows direct input of the view rotation matrix (by row), if it has been previously calculated by another program. An error message will be produced by a matrix for which the determinant is not within reasonable rounding errors of +/-1.
rotations:
(XROT xr)(YROT yr)(ZROT zr)(LROT lr x y z)
(OROT or x y z)(PROT pr x y z)
(BROT atom_name1 atom_name2 br)
Examples of VIEW instructions:
VIEW MIN
VIEW YO
VIEW AFACE ZROT 15
VIEW DIRECTION 0.25 0.25 1
VIEW LINE PT1 BR1
VIEW BISECT C1 C2 C3
VIEW PERP N1 CU1 N2
VIEW LINE C1 C2 ZROT 90 YROT -30
VIEW CURRENT BROT C1 C2 -45
The atomic parameters (including unit cell parameters, symmetry and coordinates) for a given structure may be entered in one of the following ways:
a - The STANDARD PARAMETER FILE Format (SPF).
b - SHELXL-97 and SHELXS-86 type Format
c - Simple Coordinate Format and Angstrom data
d - CIF-style (Restricted)
The SPF-file is card image oriented. The first four characters on a card specify the nature of the data that follow on that card. Data that are not needed for the current program are simply skipped. All data are read free format. All input data are transformed to upper case except the text on the TITL card.
Note: A card image may be continued on the next one by putting an '=' sign on the one to be continued. This does not apply for a TITL card.
This text is used for various titleing purposes. It may be overridden at any time by another TITL instruction. This card is optional.
Optional wavelength and cell parameters in Angstroms and degrees respectively. No CELL card should be given when Angstrom data input is intended (see ANGSTROM instruction). This card should preceed any fractional coordinate data.
Space group symbol. See Appendix-III for more details. Space group P1 is assumed when no symmetry is specified.
First parameter specifies the Bravais lattice type and the second whether the lattice is acentric or centric. See Appendix-III for details.
See appendix-I for details.
This specifies the positional parameters, the population and their estimated standard deviations. The atom_name should conform some rules in order to be acceptable since it is interpreted. The first one or two characters should correspond to an element name known to the program. The number of characters of the element type and that of the attached digital number cannot exceed 4.
Acceptable labels are: Ag Zn(2) C(2A) Fe1b
Note: QW is equivalent to O and Q1 is equivalent to C1.
This optional instruction may be used to transform the data to a new unit cell. The first nine data items are the elements of the matrix that describes the transformation of the cell axes ( = new axis in terms of old ones). The origin may be shifted over the vector (sh1,sh2,sh3). Related matrices are used to transform the atomic coordinates. The TRNS instruction should be given before any ATOM card is read and before the CELL card when it is to be transformed as well. The space group symmetry is not transformed and should apply to the transformed data.
ATOM cards may be as: C1 1.123 1.456 1.789
CIF-DATA FORMAT
A restricted format CIF-DATA file (such as produced by SHELXL-97) is acceptable.
EXAMPLE: The symmetry for space group nr. 19 (P212121) may be specified either as:
LATT P A
SYMM X,Y,Z
SYMM 1/2 + X, 1/2 - Y, -Z
SYMM -X, 1/2 + Y, 1/2 - Z
SYMM 1/2 - X, - Y, 1/2 + Z
or
LATT P A
SYMM 1/2 + X, 1/2 - Y, -Z
SYMM -X, 1/2 + Y, 1/2 - Z
or
SPGR P212121
or
SPGR P21 21 21
A LATT card should precede any SYMM card in order that the symmetry
arrays are initialized to either, by default, a primitive non-
centrosymmetric lattice or to the specified lattice type:
(P/A/B/C/I/F/R) and (A)Centric type (A/C).
The general equivalent positions should be given as specified in International Tables and should have the centre of symmetry at the origin, in the case that the space group is centrosymmetric. The symmetry operation SYMM X,Y,Z is always implicitly assumed as the first symmetry operation and needs not be given although any redundancy in the symmetry input will be ignored.
Note: Rhombohedral lattice types (in hexagonal setting) can be specified in two ways: Thus space group R3 may be generated with:
LATT P A
SYMM -Y, X-Y, Z
SYMM 1/3+X, 2/3+Y, 2/3+Z
or
LATT R A
SYMM -Y, X-Y, Z
The same space group on rhombohedral axes should be specified as R3r.
or
LATT P A
SYMM Y,Z,X
The translation part may be specified either as a ratio or as a real (e.g. 1/4 or 0.25).
Monoclinic-b is taken as the standard setting for monoclinic space groups. Other settings are to be specified by the full space group name: e.g. P112 for the monoclinic-c setting of P2.
Non-standard orthorhombic settings such as space group A2aa may be handled by specifying Ccc2.-cba on the SPGR card (see International Tables Vol A). In fact the program automatically modifies the input line accordingly for non-standard settings (see table below). The standard setting symmetry is than transformed accordingly.
Note: Symmetry may also be presented in the SHELX-76 style. However a LATT card should always be supplied since the default symmetry of PLUTON is always P1 whereas SHELX defaults to P-1.
For compatibality reasons with other programs, a space group name such as P41212 may also be specified as P 41 21 2.
PLATON HOMEPAGE
{kind=link}
{kind=link}
{kind=link}
{kind=link}