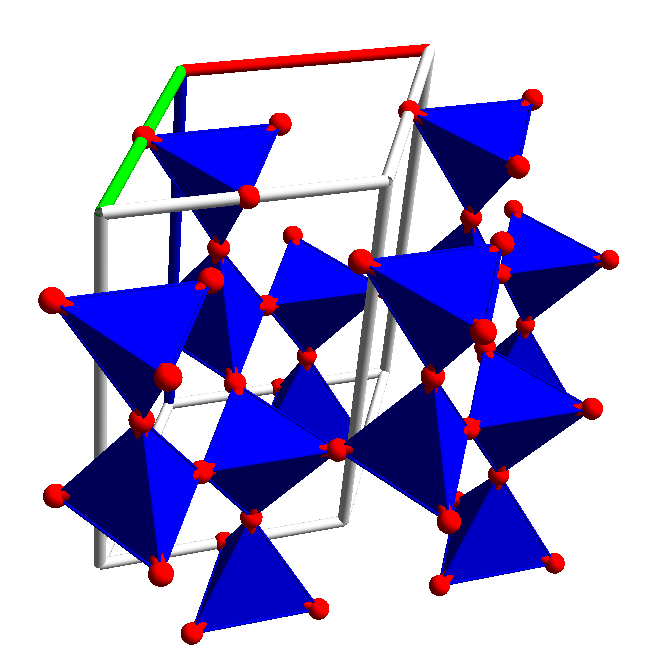
a-quartz: If you follow the procedure used in the rutile example using the quartz.ins file and pack ranges (-1 2), (-1 2) and (0 2) and tetrahedra about the Si atoms the Pogl view should be as shown here.
b-cristobalite:
The space group is number 227 Fd-3m and it has more than 99 symmops. This is
an idealized structure with the oxygens on the Si-Si line.
Cristobalite and trydimite are oxygen widened cubic
and hexagonal diamond structures respectively A good description of this and
other structures is given in "Inorganic Structural Chemistry"
by Ulrick Muller.
Start
Oscail and point it at the cristobalite.ins file in \ortex.

There are isolated atoms and "missing atoms" in this picture. To restore the "missing atoms" exit Ortex and the message

is displayed. Click OK and in Oscail select Use.LIST/Use LIST.INS. This will switch the jobname to TEST
Click ![]() (Save As Ptest) and
(Save As Ptest) and ![]() (Switch to Ptest). The Jobname is now set to Ptest and the space group to P1.
Run Ortex and select defaults.
(Switch to Ptest). The Jobname is now set to Ptest and the space group to P1.
Run Ortex and select defaults.

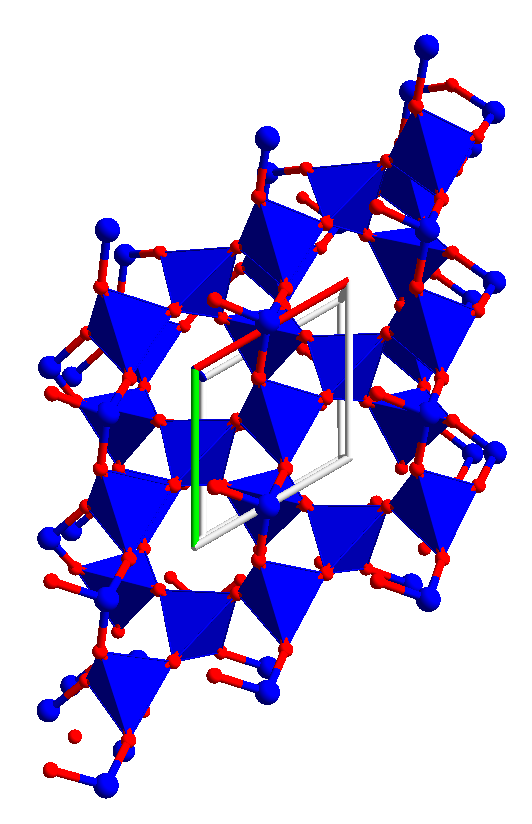
There are now no missing or isolated atoms. It is a good idea to narrow the pack range further at this stage to (-.25 1.25), (-.25 1.25) and (-.25 1.25) and exit Ortex. Run Pogl and with tetrahedra about each Si and 0.25 ambident light you should get

b-tridymite: Using the tridymite.ins file and a pack range of (0 2), (0 2) and (0 2) gives enough of the lattice to show the relationship to hexagonal diamond. The oxygens are in idealized positions.
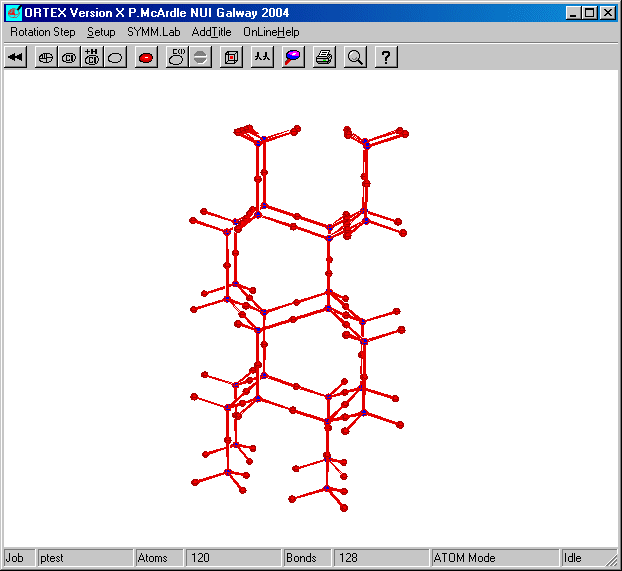
Using a smaller pack range gives