July 26, 2005
DRAWxtl V5.1
A program to make ball-and-stick, or polyhedral crystal structure drawings
by
Larry Finger, Martin Kroeker and Brian Toby
This program reads a basic description of the crystal structure, which includes unit-cell parameters, space group, atomic coordinates, thermal parameters or a Fourier map, and outputs a geometry object that contains polyhedra, planes, lone-pair cones, spheres or ellipsoids, bonds, iso-surface Fouier contours and the unit-cell boundary. Unlike earlier versions, the program now contains a graphical-user interface (GUI) that renders the structure object using a series of openGL calls using the glut-programming interface, and provides the capability to adjust many of the parameters and see the effect on the drawing in real time. As before, scene description files for the popular POV ray-tracer program, or a Virtual Reality Modeling Language (VRML) file are produced. The POV scene file can be used as is, but it can also be edited to make use of the more sophisticated features of POV like transparency, backgrounds or light effects. The VRML version is suitable for use with WWW browsers such as FireFox, and can be used to make displays suitable for interactive examination by users. Source URL’s for this and associated programs are given in Appendix A. The original version of this program was a module for the visualization software of Advanced Visual Systems, Inc.; however, that program has not been maintained for several years. That code was written by LWF (Larry.Finger@lwfinger.net), Geophysical Laboratory, Carnegie Institution of Washington, 5251 Broad Branch Road, N.W., Washington, DC 20015-1305. The original POV and VRML modifications were proposed and coded by MK (martin@ruby.chemie.uni-freiburg.de), Institut für Organische Chemie, Technische Hochschule Darmstadt, D-64287 Darmstadt, Germany. Addition of the Fourier-contour code was accomplished by Brian Toby (brian.toby@nist.gov), NIST Center for Neutron Research, Stop 8563, National Institute of Standards & Technology, Gaithersburg, MD 20899-8563.
Program Features:
DRAWxtl can make ball-and-stick, polyhedral, or mixed diagrams, with atoms represented as spheres or thermal ellipsoids. For the latter type, anisotropic coefficients can be input as Uij, Bij, or b ij, depending upon what is available. Polyhedral diagrams can be made with polyhedra of any desired shape, not just tetrahedra or octahedra. For zeolite drawings, it is also possible to make the 'polyhedron' of tetrahedral ions about the center of the cages.
The program can read structural input from CIF, CSD (the Cambridge Structure Database), GSAS, SCHAKAL, and SHELX formats, as well as its own native format. In this way, the crystallographic data for complicated structures do not have to be retyped. Fourier maps may be input in the ‘grd’ format from GSAS, the ‘stf’ format of JANA2000, the ‘w2k’ from WIEN2K, the ‘vsp’ format from VASP, or the ‘flp’ format of FullProf. Alternatively, if structure factors are available in CIF format, DRAWxtl will compute the Fourier map.
The program also outputs a table of bond distances from each of the atoms in the asymmetric unit to all other atoms. All symmetry operators are used in this calculation, with distances less than a specified limit (defaults to 3.5 Å) written to the listing file. This output can be used to check that input parameters are valid. For anisotropic atoms, the eigenvalues (RMS amplitudes) and eigenvectors for the ellipsoids are computed and listed.
This program is written in highly portable C++ and will run on a wide variety of machines including 386- to Pentium-based PC's, Macintoshes, high-end workstations, and Linux systems. The only requirements are a graphical-windows based system, a floating-point coprocessor, a C++ compiler, and libraries for openGL, and FLTK (The Fast Light Tool Kit) V1.1.6. The developers use Microsoft Visual C 6.0 for Windows and gcc and g++ for Linux. The program requires a display with at least 800 ´ 600 resolution. N.B. For those people that do not wish to use a windowing system, we have an updated, non-GUI, version available. Please check http://www.lwfinger.net/drawxtl for details.
DRAWxtl is open-source software, and is free to anyone. We hold copyright on the code, and like any other piece of intellectual property, we ask that you respect our rights.
Program Operation:
As in previous versions that were written for command-line input, a text file of instructions controls the drawing produced. In this version, however, the structure file can be changed by the widgets of the main and secondary screens, and the effect of such changes shown immediately on the main screen of the program (Fig. 1). As in earlier versions, the program produces the POV and VRML output files, which are then read by the companion renderers/viewers to produce the hard copy output, or files suitable for viewing with a Web browser. New with V5.1 is the ability to go directly from the main screen to an Encapsulated Postscript (EPS) file; thereby bypassing the external hard-copy program step for users that do not have POV installed.
Fig. 1: Screenshot of main screen for DRAWxtl.
|
|
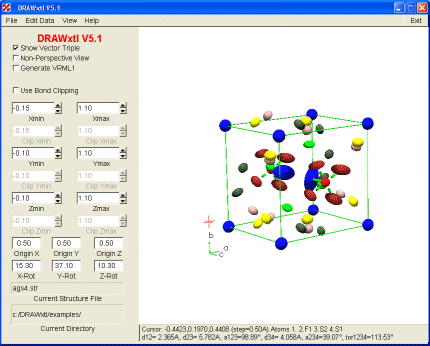
Another new feature in V5.1 and later is the graphics cursor shown in the lower, left rear of the diagram above. Its position in fractional coordinates is shown on the line below the drawing. The various bond distances and angles and the torsion angle are shown for the last 4 selected atoms. To aid in atom selection, the following keyboard shortcuts have been implemented:
- ‘C’ or ‘c’ - turn the cursor on. Each succesive press reduces the size of the steps. When the size is reduced below 0.01 A, the cursor is turned off.
- ‘P’ or ‘p’ - place the graphics cursor on the atom nearest the mouse cursor.
- ‘A’ or ‘a’ - place the cursor at the position of the atom nearest the cursor.
- ‘M’ or ‘m’ – move the cursor to the min/max in the electron-density contours.
- ‘x’, ‘y’, ‘z’ – move the cursor positive in the along the x-, y- or z-axis.
- ‘X’, ‘Y’, ‘Z’ – move the cursor negative in the along the x-, y- or z-axis.
- ‘L’ or ‘l’ - label the atom at the cursor position.
- ‘B’ or ‘b’ – Place the bond distance between atoms 1 and 2 in the list.
- Hold leftmouse – rotate the graphics object
- Hold rightmouse – zoom in/out on the object
- Hold middlemouse (both on 2-button mouse) – pan motion
- Shift/leftclick – drag labels or the triple vector to a desired position.
- Ctrl/leftclick – remove the object at the location of the mouse cursor.
With the widgets located on the main screen, the user can select how much of the structure to portray, where to place the origin, and what orientation to view. Dragging the mouse in the graphics window can also change the latter parameters. With the menu items at the top of the screen, the user may select the structure to be displayed, edit the various parameters, view the POV file or listing output, set some POV configuration parameters, or view the various help files. Fig. 2a shows the screen used to edit polyhedron or plane parameters and 2b shows the screen for bonds. Similar screens exist for ellipsoids, spheres, and general parameters. For those changes not addressed by these screens, the user also has the ability to edit the original or revised ‘str’ file.
|
Fig. 2a. Polyhera/Planes edit screen |
2b. Bond edit screen |
|
|
|
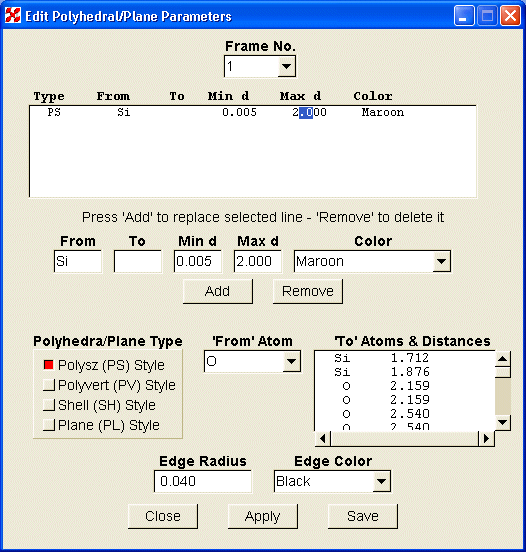
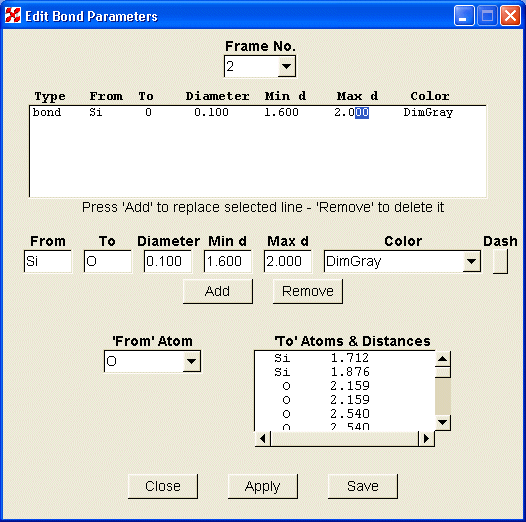
The edit screens show the current object descriptions in the upper window, where the parameters can be modified, either by direct editing, or by double clicking on a line of the window. The data will be transferred to the boxes below, where changes may be made. When all boxes are filled, the ‘Add’ and/or ‘Delete’ buttons will be made active. New objects can be created using the lower part of the screen. After selecting the central atom with the ‘From Atom’ combo box and using the ‘Polyhedra/Plane Type’ radio buttons to select the type of object, the user highlights one or more characters in the line containing the coordinating distance to be selected. The parameters will be transferred to the middle row of boxes. After any necessary changes are made in that box, press the ‘Add’ button to create a new line. When either the ‘Apply’ or ‘Save’ button is pushed, the new parameters will be applied to the drawing. When the structure has multiple frames, the "Frame No," combo box is used to select the frame that is to be modified.
Input Instructions for DRAWxtl:
In this program, color is represented in symbolic form, and must be one of the color names from POV’s "colors.inc" file, which may be modified to define custom colors. The standard color names and associated RGB values are listed in Table I, and sorted by RGB values in Table II. Any of these colors may be made transparent by appending the phrase ‘filter xx’ after it, where ‘xx’ is a number from 0.0 to 1.0. The larger the value of ‘xx’, the more transparent will be the entity. Each line of the input file is preceded by a character sequence that describes the type of information, as follows:
arrow ‘xp’ ‘yp’ ‘zp’ ‘xc’ ‘yc’ ‘zc’ ‘length’ ‘diameter’ ‘color’
defines the position in fractional coordinates (xp, yp, xz) of the nuclear cell, the components (xc, yc, zc) of the spin vector, and the length, diameter, and color of the arrows The reference direction for xc is parallel to direct space a, yc is parallel to (a ´ b) ´ a, and the reference direction for zc is perpendicular to xc ´ yc. The only symmetry elements used in placing arrows are the translations described by the mag_trans command below.
atom ‘name’ ‘number’ ‘x’ ‘y’ ‘z’
defines the atoms. The 1- to 4-character name will be used on the commands that describe the objects to be created, the number is to identify which atom of this type is referenced, and ‘x’ ‘y’ ‘z’ are the fractional coordinates in the unit cell. If the atom is located on a special position where the coordinate is given by a fraction such as ¼, the string ‘1/4’ may be entered rather than 0.25.
axislines ‘width’ ‘color’
defines the width and color of the lines that depict the principal axes of ellipsoids. The color defaults to dark gray (Gray20). If this command is not given, any ellipsoids will be drawn with principal axes of 0.00015 times the overall scale factor, which should normally be appropriate.
background ‘color’
sets the color of the background of the graphical views. The default color is white.
betaij ‘name’ ‘number’ ‘b 11’ ‘b 22’ ‘b 33’ ‘b 12’ ‘b 13’ ‘b 23’ ‘color’
defines the anisotropic thermal coefficients for an atom and ‘color’ of the ellipsoid. The ‘name’ and ‘number’ should correspond to the atom input described above. For this form, the temperature factor is given by T = exp{-S jS khjhkb jk}. In the POV version of the program, the principal ellipses are drawn in black. The ellipses will, of course, be invisible if the ellipsoid is also black.
bij or Bij ‘name’ ‘B11’ ‘B22’ ‘B33’ ‘B12’ ‘B13’ ‘B23’ ‘color’
defines the anisotropic thermal coefficients for an atom and the color of the ellipsoid to be drawn. The ‘name’ and ‘number’ should correspond to the atom input described above. For this form, the temperature factor is given by T = exp{-0.25S jS khjhkBjkaj*ak*}, where aj* and ak* are reciprocal lattice constants.
bond ‘name1’ ‘name2’ ‘radius’ ‘min length’ ‘max length’ ‘color’,
where ‘name1’ and ‘name2’ indicate the types of atoms to be connected by a bond, ‘radius’ is the radius of the resulting cylinder, and the minimum and maximum lengths are given in the same units as the unit cell.
box ‘radius’ ‘color’
defines the radius and color of the cylinders that form the unit cell boundary. If ‘radius’ is 0.0, plotting of the unit cell is suppressed. The radius of the cylinders will be scaled with the size of the drawing. The default size is 0.02.
cell ‘a-length’ ‘b-length’ ‘c-length’ ‘alpha’ ‘beta’ ‘gamma’
unit-cell lengths and angles. If no angles are listed, they are assumed to be the fixed values for the symmetry class.
clip ‘xmin’ ‘xmax’ ‘ymin’ ‘ymax’ ‘zmin’ ‘zmax’
defines a-,b-,c- clipping range in fractions of the axes. Any bonds extending beyond these limits will be cut off at half-length. This command is to be used in conjunction with the pack keyword to produce 'dangling' bonds in the display of framework structures.
cutout ‘color’ (used only for POV and openGL)
sets the POV generation of thermal ellipsoids to have one octant removed, as in the program ORTEP. If this command is not given, all ellipsoids will be complete. The color is for the planes that describe the edges of the cutout
dash ‘name1’ ‘name2’ ‘radius’ ‘min’ ‘max’ ‘color’
where ‘name1’ and ‘name2’ indicate the types of atoms to be connected by a dashed bond, ‘radius’ is the radius of the resulting cylinder, and the minimum and maximum lengths are given in the same units as the unit cell.
depthcue ‘depth’ (used only for POV)
defines the extent to which the size of polyhedral edges is increased as the edge is closer to the viewer.
edges ‘radius’ ‘color’
defines the thickness and color of cylinders along the edges of polyhedra that may be used to emphasize the faces. The radius of these cylinders will also be scaled with the size of the drawing. By default, black edges of size 0.02 will be drawn.
ellipcolor ‘name’ ‘number’ ‘color’
defines the color for ellipsoids when the thermal ellipsoid information has been read from a CIF, CSD, GSAS, SCHAKAL or SHELX import or inline file. The ‘name’ and ‘number’ must match the identification information in the input file. The parameter ‘number’ may be an asterisk (*) to indicate all atoms with that name. In addition, these input lines must be after the import or inline command.
ellipsoids ‘probability’
sets the size of the ellipsoid such that that fraction of the electron density is contained within the bounding surface. Use either 0.50 or 50 to get the standard (default) 50% ellipsoids.
finish ‘ambient’ ‘diffuse’ ‘specular’ ‘roughness’
defines parameters for the POV lighting functions that are applied to all surfaces. Suggested values are 0.7 0.3 0.08 0.01 to reduce the harsh contrasts that can result from the default material properties in POV.
frame ‘comment’
similar to ‘end', marks the division between two sets of input that are to be superimposed in a single output file. Each frame must have a complete set of ‘atom’ lines, and lines describing the objects such as bonds, polyhedra, spheres, etc. to be created in this frame. Distinct packing ranges and space grpups are optional. A prime example of this command would be to draw structures with adsorbed molecules with symmetry lower than the cage in which it resides. Another option is to draw ball-and-stick and polyhedral pictures in side-by-side unit cells. All other parameters are global and apply to all frames.
import ‘file type’ ‘filename’ ‘phase number(for GSAS only)’
causes the program to read structural information from an external file. ‘file type’ defines the format of that file, and ‘filename’ is the name of the file. Import filters have been written for GSAS, SHELX, and CIF formatthe CIF, FDAT (Cambridge Structure Database), GSAS, SCHAKAL, and SHELX formats. For GSAS format, the number of the phase should also be given. From these files, the atomic coordinates, thermal parameters, and unit cell will be read. For the GSAS and CIF formats, the space group will alsounit cell, and space group (CIF, FDAT, GSAS and SCHAKAL formats only) will be read. To set colors for the ellipsoids, use the ellipcolor command. You must also use the ellipsoids command to get ellipsoids displayed. In files that are imported, atom names of the form Si3A will have atom names of SiA with a number of 3.
inline ‘file type’
is similar to import, except that the ‘foreign’ input information is included in the DRAWxtl input file. This form works for CIF, FDAT, SCHAKAL and SHELX data. To set colors for the ellipsoids, use the ellipcolor command. You must also use the ellipsoids command to get ellipsoids displayed.
labelsize ‘size’
changes the relative size of the labeltext entries. The default value is 1.0.
labeltext ‘x’ ‘y’ ‘z’ ‘text string’
plots the given string at the specified position given in fractional coordinates.
list ‘maxdist’
causes the program to list bond distances up to ‘maxdist’ in the preliminary scan. If this command is not given, ‘maxdist’ defaults to 3.5 Å
lonepair ‘name’ ‘number’ ‘distance’ ‘radius1’ ‘radius2’ ‘color’
creates the specified number (either 1 or 2) of cones representing free electron pairs extending from atom 'name', where 'distance' is the length of the cone, ‘radius1’ is the size of the tip, and ‘radius2’ is the size of the spherical end cap.
lookat ‘u1’ ‘u2’ ‘u3’ ‘v1’ ‘v2’ ‘v3’
causes the program to select an orientation such that vector u is towards the viewer, and the projection of vector v is vertical. This command overrides any view command, or the -v switch on the command line.
mag_trans ‘Aa’ ‘Ab’ ‘Ac’ ‘Ba’ ‘Bb’ ‘Bc’ ‘Ca’ ‘Cb’ ‘Cc’
describes the relationship between the magnetic and nuclear unit cells. In this notation, the upper-case letter states which of the magnetic axes is being described, and the lower-case letter corresponds to the nuclear cell axis. This matrix defaults to the identity.
magnification ‘factor’
sets the factor to modify the overall scaling is case the automatic value is not correct. This command matches the -m command line switch.
mapcalclimits ‘xmin’ ‘xmax’ ‘ymin’ ‘ymax’ ‘zmin’ ‘zmax’
describes the region of direct space (in fractional coordinates) for which the map has been calculated. Map types that are self documenting such as FullProf and JANA2000 do not need this line. For other types, 0 to 1 in all three directions will be assumed.
mapcontour ‘level’ ‘style’ ‘color’
defines a new contour at 'level'. The style can be either 'mesh' or 'solid', and the color is set by 'color'
mapread ‘maptype’ ‘filename’ ‘calctype’
reads a Fourier map of type 'maptype' from the file named 'filename'. At present, GSAS-style (maptype = grd), JANA2000-style (maptype = stf), WIEN2k (maptype = w2k), VASP (maptype = vsp), and FullProf (maptype = flp) maps are read. If a Shelx/Cif-style Fo/Fc file (maptype = fcf) is given, the electron density is calculated during the initial read, which may take a few minutes. Both A/B and Fo/phi data formats (Shelx commands LIST 3 and LIST 6) are supported. Parameter ‘calctype’ is needed only for maptype=fcf, and specifies which type of map is to be calculated. The valid options are Fo, Fc, Fo-Fc, 2Fo-Fc, and Fo2 (i.e. a Patterson map).
mapregion ‘xmin’ ‘xmax’ ‘ymin’ ‘ymax’ ‘zmin’ ‘zmax’
describes the region of direct space (in fractional coordinates) that the map is to be displayed in the output. If not entered, these values default to the values given under 'mapcalclimits'.
molcomp ‘dist’
causes any incomplete molecules in the display box to be completed. The value of ‘dist’ defines the maximum intramolecular distance. Caution: If this distance is greater than any intermolecular distance, or if the material is not molecular, the display list will overflow.
nolabels
removes all axis labels from the output diagrams.
noshadow
causes objects in the POV file not to cast shadows.
origin ‘xcenter’ ‘ycenter’ ‘zcenter’
defines center of view box in crystal coordinates (defaults to 0.5 0.5 0.5).
orthographic
causes the camera to be changed from the normal perspective view to an orthographic view for VRML and removes all perspective from POV diagrams.
pack ‘xmin’ ‘xmax’ ‘ymin’ ‘ymax’ ‘zmin’ ‘zmax’
defines a-,b-,c- plotting range in fractions of the axes, similar to the Pluto (Motherwell & Clegg 1978) PACK RANGE command (this is especially useful for highly oblique cells, where the orthorhombic view box does not always give satisfactory results).
phong ‘value’ ‘size’ (used only for POV)
defines the amount of Phong highlighting on spheres and ellipsoids. The ‘value’ ranges between 0.0 and 1.0, where 0.0 gives no highlight, and 1.0 causes complete saturation at the center of the highlight. The ‘size’ ranges from 1.0 (very dull) to 250 (highly polished). The default quantities are 0.1 and 1.0, which gives a large, dull highlight. If ‘value’ is 0.0, the image can be rendered much more quickly.
plane ‘name’ ‘length’ ‘color’
defines the center of a plane group, such as CO3, that is to be drawn in a structure, where ‘name’ is the name of the atom at the center and ‘length’ is the maximum distance to coordinating anions.
polyedge ‘name’ ‘radius’ ‘color’
defines the thickness and color of cylinders used to emphasize the faces along the edges of polyhedra for atom ‘name’. The radius of these cylinders will also be scaled with the size of the drawing.
polysz ‘name’ ‘length’ ‘color’
defines a polyhedron, where ‘name’ is the name of an atom at the center of a polyhedron and ‘length’ is the maximum length of distances to atoms that are to be considered as the vertices of the polyhedron. The polyhedra can be of any desired complexity - more than tetrahedra or octahedra can be drawn. For polyhedra with both upper and lower limits (which might be desirable for intermetallic compounds), use the 'shell' command.
polytolerance ‘length’
polylimit ‘length’
polyfudge ‘length’
modifies the internal limit for the deviation of vertices from the common plane. While the default value (0.1) will always generate correct drawings, it may sometimes be desirable to increase it to create idealized views of nearly symmetrical polyhedra that would otherwise show creased surfaces.
polyvert ‘name1’ ‘name2’ ‘length’ ‘color’
defines polyhedra in the manner of the ‘polysz’ command, except that the polyhedron around the ‘name1’ atom will only include atoms of type ‘name2’.
rem, REM
Any line preceded by this command is ignored.
shell ‘name’ ‘length1’ ‘length2’ ‘color’
defines a polyhedral shell, where name is the name of an atom at the center of a polyhedron and length1 and length2 are the mimimum and maximum lengths of distances to atoms that are to be considered as the vertices of the polyhedron. The polyhedra can be of any desired complexity, and can be stacked as desired.
slab ‘a’ ‘b’ ‘c’ ‘alpha’ ‘beta’ ‘gamma’ ‘xoff’ ‘yoff’ ‘zoff’ ‘xrot’ ‘yrot’ ‘zrot’ ‘flag’
defines a (possibly oblique) cutout box of the specified axis lengths and angles that is offset by xoff,yoff zoff from the origin of the structure and rotated at angles xrot yrot zrot relative to it. If flag is set to 1, any part of the structure outside the box is deleted. If flag is 2, the outline of the box is overlaid on the unchanged image to allow accurate placement of the cutout box.
spgp, sgrp, or spgr ‘symbol’
Space Group name consisting of the Bravais lattice symbol (must be upper case) followed by a space, the elements parallel to the first axis followed by a space, etc. Examples are I 41/a m d, P 21/n, I a 3 d, P b n m, etc. The generators will always select the origin choice with a center of symmetry at the origin. Furthermore, all monoclinic cells will have the unique axis parallel to the b axis, unless the full symbol is used, i.e. P 1 1 21/n describes a monoclinic cell with c as the unique axis. N.B. Rhombohedral space groups must be represented in the hexagonal form.
sphere ‘name’ ‘radius’ ‘color’
where ‘name’ is a one- or two-character symbol of the atom type, ‘radius’ is the radius of the sphere in Angstrom, and ‘color’ is the color of the sphere to be drawn.
title, titl ‘descriptive material’
General description of the structure - this line may appear anywhere in the file, but is generally first.
uij, Uij ‘name’ ‘number’ ‘u11’ ‘u22’ ‘u33’ ‘u12’ ‘u13’ ‘u23’ ‘color’
defines the anisotropic thermal coefficients for an atom and color of the ellipsoid. The ‘name’ and ‘number’ should correspond to the atom input described above. For this form, the temperature factor is given by T = exp{-2p 2S jS khjhkbjkaj*ak*}, where . aj* and ak* are reciprocal lattice constants.
vectors
turns on the orientation vector triple at a corner of the diagram.
view ‘xrot’ ‘yrot’ ‘zrot’)
where ‘xrot’, ‘yrot’ and ‘zrot’ are view rotation angles in Cartesian space. These values correspond to a rotation of ‘xrot’ about the x axis, followed by a rotation of ‘yrot’ about the new y axis, and, a rotation of ‘zrot’ about the new z axis.
vrml1 (used only for VRML)
causes the output VRML file to have the older, VRML1 syntax for compatibility with some older viewers that are not VRML97 compliant.
vrml2 or vrml97 (used only for VRML)
These commands do nothing, but are kept for compatibility with old data files. VRML97 format is now the default. A description of some of the VRML97 features is given in Appendix B. Do NOT use this form unless your VRML viewer is VMRL97 compliant!
xyzoff ‘u1’ ‘u2’ ‘u3’
causes all atom coordinates to be shifted by -u. This command is used whenever the origin defined for a structure does not conform to the standard origin selected by the space-group generator.
end, END
is the last line of a file that is processed. Any information past this point will be ignored.