Crystal Growth, Evaluation and Handling
Alexander J. Blake
School of Chemistry, The University of Nottingham, Nottingham UK NG7
2RD
This document is best viewed with
Netscape or Internet Explorer
6.0. (or later)
Introduction
Protect your crystals
Survey of methods
Solution methods
Cooling
Convection
Concentration
Solvent diffusion
Vapour diffusion
Reactant diffusion
Seed crystals
Sublimation
Fluid phase growth
Solid-state synthesis
General comments
Evaluation
Microscopy
X-ray photography
Crystal mounting
Air-sensitive crystals
Crystal alignment
References
Crystal growing tips
on the Web
Whether growing crystals or giving advice
to someone else trying to do so, it is vital to remember that
the quality of the crystal from which diffraction data are acquired
is generally the main determinant of the final quality of the structure.
The effects of a sub-optimal crystal will propagate through data
collection, structure solution and refinement to affect the quality
of the final structure, in which unsatisfactorily high uncertainties
may limit useful comparison and discussion. It may be difficult
or mounti
ble to get such a structure published. The term recrystallisation has two related meanings but there
are crucial differences between these. In the synthesis
and purification of compounds , the aim is to maximise
purity and yield, although these can be mutually exclusive. The
material is often precipitated very rapidly ( ca. 1 second),
resulting in microcrystalline or virtually amorphous products that
are useless for conventional single crystal work. For diffraction
work the object is to obtain a small number (one may
do) of relatively large (ca. 0.1–0.4 mm) single crystals.
As long as this is achieved, yield is irrelevant and purity is likely
to be enhanced. To this end crystals should be grown
slowly, taking from minutes to months depending on the system. To understand
why this is important, visualise the process of growth at a crystal
surface. The greater the rate at which molecules arrive at the surface,
the less time they have to orient themselves in relation to molecules
already there: random accretion is more likely, leading to crystals which
are twinned or disordered. Suitable growth conditions include the absence
of dust and vibration: if these are present they can lead to crystals
which are small or non-singular.
Before you start trying to grow crystals you need to think about how they will be handled. Rather obviously,
they will need to be extracted from the vessel they grew in without suffering any damage. Less obviously,
some containers make this easier than others: an oversized one like a 250 ml round-bottomed flask
makes the procedure of finding and removing a small crystal unnecessarily difficult.
At the other extreme, a container with a small aperture that will not admit a narrow spatula or pipette is also
troublesome. You should avoid screw-top or other containers which narrow near the top, as the "shoulders"
prevent easy removal of the crystals: a small vial with straight walls works best.
Many crystals lose solvent on removal from the solution in which they have grown (mother liquor).
Although the envelope
of the crystals may appear intact, any significant loss usually renders them useless for structural
analysis. This is particularly common for crystals grown from chlorocarbon solvents like dichloromethane
and chloroform, but can affect crystals grown from almost any solvent, water included. If a crystal loses
solvent and then does not behave well on the diffractometer, there is no way to know whether the original
crystal was unsuitable, or whether the poor diffraction was solely due to solvent loss. For this reason
we strongly recommend submitting crystals under mother liquor whenever possible. There are other good
reasons for doing this - see below.
(A)
Solution methods
These are by far the most flexible and
widely used. They are suitable for use with molecular compounds
that are the subject of most crystal structure determinations.
The use of solvents means that crystals can grow separately
from each other. It is therefore important not to let a solution dry
out, as crystals could become encrusted and may not remain single.
When choosing solvents remember the general rule that
"like dissolves like": look for a solvent that is similar to the
compound (in terms of polarity, functional groups, etc) and an
anti-solvent that is dissimilar to it in order to reduce its solubility
(see below). Information about solvents is available from several
sources (e.g. Handbook of Physics and Chemistry , chromatographic
elution data), and the process of synthesising and purifying a compound
will often confer a knowledge of suitable solvents. Mixing solvents
allows manipulation of solubility: a mixture of solvent A (in which
a compound is too soluble) and anti-solvent B (in which it is not
sufficiently soluble) may be more useful than either alone. If crystals
grown from one solvent are poor, try different solvents or mixtures
of solvents. Solution
methods can be extremely flexible: a number of crystallisations,
differing in the proportions of solvents A and B used, can be set
up to run in parallel. If a particular range of proportions appears
to be more successful in producing crystals it can be investigated
more closely by decreasing the difference between successive mixtures
of A and B.
It is important that any vessels used
for crystal growth should be free of contaminants. Older
containers also tend to have a large number of scratches and
other surface defects, providing multiple nucleation points
and tending to give large numbers of small crystals. Two factors
which favour the formation of twinned crystals are the presence
of impurities and uneven thermal gradients. Conversely, if
the inner surface of a container is too smooth this may inhibit
crystallisation. If this appears to be the case, gently scratching
the surface with a metal spatula a few times may be effective. Some
of the possible variations are described briefly below, and virtually
all methods described can be adapted to accommodate air-sensitivity.
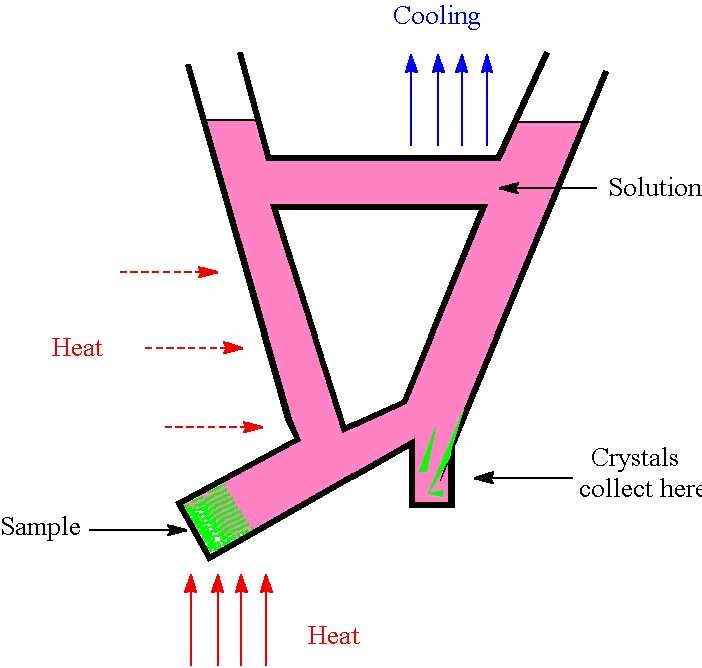
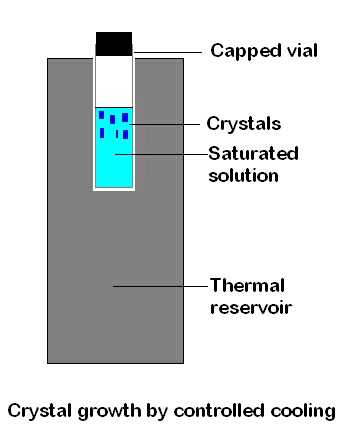
Cooling.
Either make up a hot, nearly saturated
solution and allow it to cool slowly towards room temperature
or make up such a solution at room temperature and
cool it slowly in a fridge or freezer. The cooling rate can
be reduced by exploiting the fact that the larger and more massive
an object the longer it will take to lose heat. Thus, a hot solution
in a large vessel (or in a small vessel within a larger one) will cool
relatively slowly. Similarly, a sample tube containing a solution
will cool at a slower rate if it is contained in a metal block that
was originally at room temperature, or if surrounded by an effective
layer of insulation. Cooling methods are based on the generally valid
assumption that solubility decreases with temperature. There are
rare exceptions to this ( e.g. Na 2SO4
in water) and some solubilities rise so rapidly with temperature
that it can be difficult to control crystallisation ( e.g.
of KNO3 from water). However, it is usually possible
to find a combination of solute and solvent where solubility varies
slowly and controllably with temperature.
Convection
. The aim here is to establish a temperature
gradient across the solution, so that material dissolves
in the warmer area and deposits in the colder. This gradient
can be established by various means, for example (a) allow sunlight
to shine on one part of the vessel (b) put one part of the vessel
against a cooler surface, such as a window at night (c) construct an
apparatus with low-power electrical heating elements in some sections
(see diagram). A smooth concentration gradient will give the best
results.
An apparatus (Jones, 1981) for crystal growth by exploiting convection
Concentration
.
If the volume of a solution is reduced, for example by
evaporation of a volatile solvent, the concentration of
the solute will rise until it begins to crystallise. When using
mixed solvents the poorer solvent should be the less volatile
so that the solubility of the solute decreases upon evaporation.
The rate of evaporation can be controlled in various ways, for
example by altering the temperature of the sample or by adjusting
the size of the aperture through which the solvent vapour can escape.
As solvents are frequently flammable or irritant, it is important
to work on the smallest scale possible and ensure than any vapour released
from the solution is safely dealt with. Avoid obvious hazards such
as those that will arise if large volumes of diethyl ether or other
highly volatile solvents are allowed to evaporate in a closed container
such as a refrigerator.
As noted above, it is highly undesirable to let a solution evaporate to
dryness: this will allow otherwise suitable crystals to become encrusted,
grow into an aggregate or be contaminated by impurities. The crystals may be
degraded by loss of solvent of crystallisation, especially if chlorocarbon
solvents such as dichloromethane have been used. It may prove impossible
to identify good crystals even if these are present, and extracting
them undamaged from a mass of material may prove difficult.
Apparently sealed NMR tubes which have
been forgotten at the back of a fume cupboard or fridge for
weeks or months are a fruitful source of good quality crystals:
there is in fact slow evaporation of solvent and crystals are able
to grow undisturbed. As long as the NMR tube is clean and relatively
unscratched the smooth inner surface and narrow bore provide an
excellent environment for crystal growth.
One method that combines variation of
both concentration and temperature and is especially useful
for sparingly soluble compounds is Soxhlet extraction. The
recycling of the solvent is the key factor here and crystals can
even appear in the refluxing solvent. Failing this, they normally
appear after slow cooling of the solution. There are several other
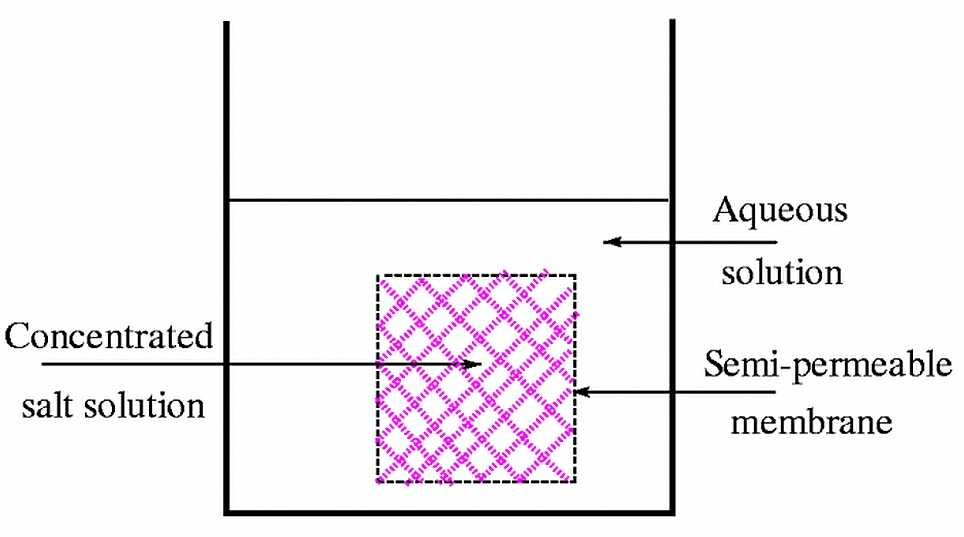
methods available to control concentration. One of these is based
on osmosis, where the solvent passes through a semi-permeable membrane
into a concentrated solution of an inert species. The resulting increase
in the concentration of the solute may lead to crystal formation.
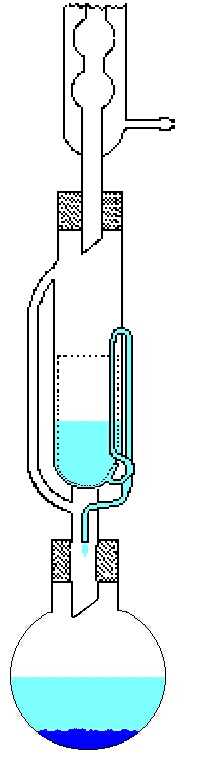 A Soxhlet extraction apparatus used for growing crystals
of poorly-soluble materials
A Soxhlet extraction apparatus used for growing crystals
of poorly-soluble materials
Use of osmosis to control concentration
Selective removal of the less volatile solvent (CH
2Cl2) from a mixture (CH2Cl2/Et
2O)
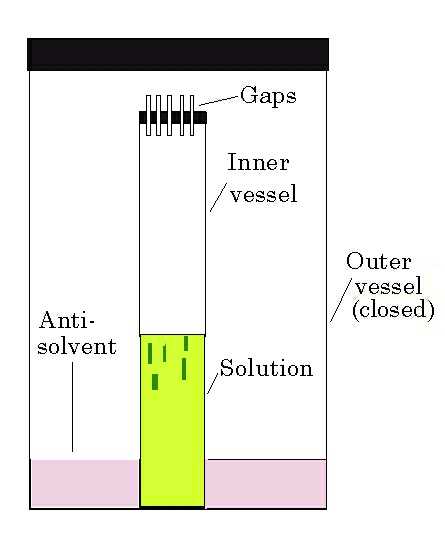
Solvent diffusion
. This method is based on the fact that
a compound will dissolve well in certain solvents ("good"
solvents) but not in others ("poor" solvents or anti-solvents),
which must be co-miscible. Dissolve the compound in the "good"
solvent and place this solution in a narrow tube. Using a syringe
fitted with a fine needle, very slowly inject the neat anti-solvent.
If it is lighter than the solution, layer it on top; if it is denser,
inject it slowly into the bottom of the tube to form a layer under
the solution. Injecting the solvent is better than running it down
the side of the tube. If the tube is protected from vibration, these
layers will mix slowly and crystals will grow at the interface. If
necessary, cooling of the tube can be used both to lower the rate of
diffusion and to reduce the solubility.
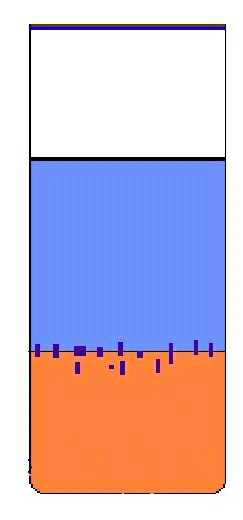 Crystal growth by solvent diffusion
Crystal growth by solvent diffusion
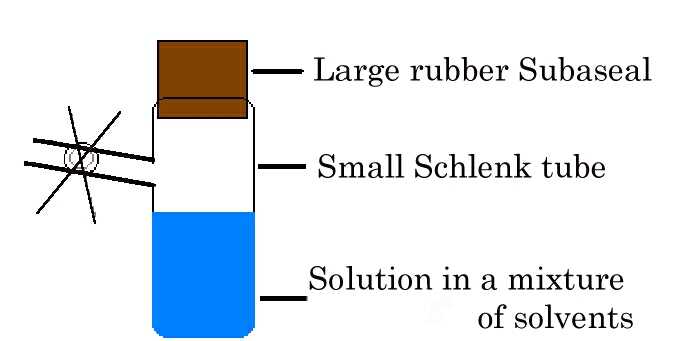
Vapour diffusion.
This method is also called isothermal
distillation. The anti-solvent diffuses through the vapour
phase into a solution of the compound in the "good" solvent,
thereby reducing the solubility. The advantages of this method
include the relatively slow rate of diffusion, its controllability
and its adaptability, for example in combination with Schlenk
techniques to grow crystals of air-sensitive samples. It is usually
worth trying vapour diffusion as it frequently succeeds where
other methods have failed. A variant on this is the hanging drop
method, principally used for the growth of crystals of proteins and
other macromolecules: the precipitant sits in a well and diffuses
slowly into a drop of solution suspended on a glass slide covering
the well.
Crystal growth by vapour diffusion
Reactant diffusion
. It is sometimes possible to combine
synthesis and crystal growth. In favourable cases crystals
may simply drop out of the reaction mixture, but the rate
of many reactions means that crystals form rapidly and are therefore
small and of low quality. If the reaction rate can be controlled
by slow addition of one of the reactants this offers one way to
overcome the problem. The best control is often achieved by controlling
the rate at which reactant solutions mix, by interposing a semi-permeable
barrier ( e.g. membrane, sinter or an inert liquid
such as Nujol) or by the use of gel crystallisation (see below).
Another variant involves placing a solid reactant at the bottom of
a tube, covering it with a solvent in which it is known to dissolve
slowly, and carefully adding an upper layer consisting of a solution
of a second reactant.
The additional time required for the solid to dissolve
reduces the rate at which reaction can occur.
Crystals of zeolites and of many other
materials with network structures cannot be recrystallised
and therefore can only be obtained from the reaction mixture.
Fine tuning of the reaction conditions and the proportions and
concentrations of reactants probably offer the only realistic ways
to control crystal size and quality.
Crystallisation from gels is an under-exploited
technique for obtaining single crystals of compounds of
low solubility.
Because the mixing of the solutions is dominated by diffusion
through a viscous medium, undesirable competing processes such as
convection and sedimentation are minimised. It is therefore possible
to establish laboratory conditions for crystallisation that closely
approximate the microgravity of space. A typical arrangement is
a U-tube half-filled with gel, with a solution of one reactant in the
top of one arm and a solution of another reactant in the other.
As gels are generally colourless it is much easier
to detect and isolate crystals of a strongly coloured product. There
are various recipes for the preparation of gels (e.g. http://www.cryst.chem.uu.nl/lutz/growing/gel.html;
H. Arend & J.J. Connelly, J. Cryst. Growth 1982,
56 , 642) and it is possible to treat gels with organic
solvents to produce versions suitable for use with hydrophobic
or moisture-sensitive compounds.
Seed crystals.
Sometimes crystallisation of a compound
gives crystals which, although otherwise of good quality,
are clearly too small for structure analysis. A small number
of these can be used as seeds by placing them into a warm saturated
solution of the compound and allowing the solution to cool slowly.
The hope here is that crystal growth will occur preferentially
at the seed to give a suitably large single crystal. A container free
of contaminants and scratches is strongly recommended here.
(B)
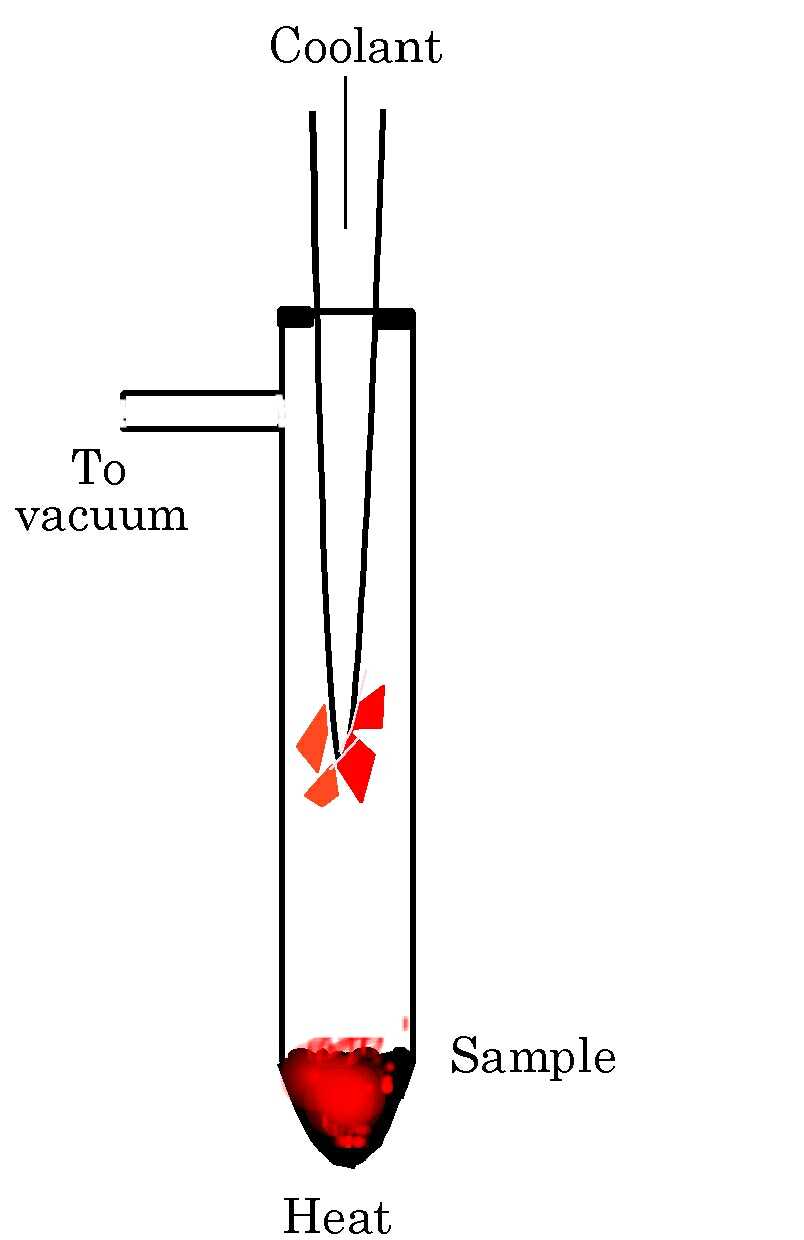
Sublimation
Sublimation is the direct conversion
of a solid material to its gaseous state. It has been harnessed
to produce solvent-free crystals of electronic materials
but it is applicable to any solid with a significant vapour pressure
at a temperature below its decomposition or melting point.
The basic experimental arrangement is simple: a closed, usually
evacuated vessel in which the solid is heated (if necessary) and
a cold surface on which crystals grow. If possible avoid heating
the solid, as lower sublimation temperatures often lead to better
crystals. If the solid sublimes too readily the vessel can be cooled.
If a compound has a low vapour pressure, sublimation can be enhanced
by evacuating the vessel or by using a cold finger containing acetone/dry
ice (‑78 o C) rather than cold water (5–10
o C).
Using sublimation to grow crystals without the use of solvents
(C)
Fluid phase growth
It is possible to grow crystals directly
from liquids or gases, often by employing in situ techniques.
Fluid phase methods encompass both high temperature growth
from melts and low temperature growth from compounds that melt
below ambient temperature. High temperature methods (Bridgman,
Czochralski, zone refining, etc) are used widely in the purification
and growth of crystals of semiconductors and other electronic
materials but are limited to compounds that melt without decomposition,
thereby excluding many molecular compounds. Moreover, it is much
more difficult to prevent unwanted phenomena such as twinning
than with solution methods, and often impossible to separate overlapping
or adjacent crystals. Liquids or gases must be contained, for
example in a capillary tube. One consequence of this is that crystallisation
conditions must be controlled to give only one crystal in that part
of the tube that will be within the X -ray beam. Once
crystals have grown it is usually impossible to separate them physically.
Unlike crystal growth from solution there is essentially only one
variable, namely the temperature of the sample. However, there are
several ways to do this and the method can be chosen to give coarse
or fine control. A typical strategy for crystal growth involves the
establishment and manipulation of a stable interface between liquid
and solid phases. With air-stable compounds that crystallise in
a fridge or freezer it is only necessary to keep them cold until they
are transferred into the cold stream of the diffractometer's low temperature
device.
(D)
Solid-state synthesis
In favourable circumstances it may be
possible to produce adequate single crystals, but microcrystalline
samples are far more typical. For example, most high T
c superconductors do not give single crystals
and their structures have been determined using powder diffraction
methods. As with the synthesis of zeolites from solution, variation
of synthetic conditions is likely to be the only route to better
single crystals.
(E)
General comments
The details of crystal growth are often
poorly understood, especially for new compounds, and it
is important not to be discouraged if initial attempts fail.
For example, microcrystalline material is not immediately
useful but it does indicate that the compound is crystalline
and that modification of the crystallisation technique could
result in larger crystals.
It is always a good idea to try a range of techniques, keeping
a detailed record of the exact conditions used and the results
obtained. This not only allows identification of the most promising
methods and conditions for the current sample but also means that
in future there will be database of procedures and their outcomes
to consult. Crystal quality improves with experience, and early
attempts often produce poor quality crystals. It is important to
continue until it is clear that no further improvement is likely.
In some cases, regardless of the method
employed, crystals either do not form or are unsuitable.
At this stage, the best way to proceed may be to modify the
compound. With ionic compounds it may be practical to change the
counterion (e.g, BF4- for PF6-,
or vice-versa ). With
neutral compounds it may be a simple matter to change some chemically
unimportant peripheral group. In one case altering a piperidine
substituent to morpholine, which merely involves changing one
remote CH2 group for an oxygen atom, led to a spectacular
improvement in crystal quality.
Once crystals have appeared it is necessary
to ascertain whether they are suitable for data collection.
Some of the methods used are extremely rapid and can save
large amounts of diffractometer time. During these procedures
take care to prevent damage to the crystals, for example by loss
of solvent after removal from the mother liquor. If spare crystals
are available, leave one or two exposed on a microscope slide and
check them regularly for signs of deterioration, using microscopy
as described below. It is vital to apply the tests outlined below
optimistically so that only crystals that
are incontrovertibly unsuitable are rejected. Any that give uncertain
indications of their quality should be given the benefit of the
doubt.
1.
Microscopy.
Visual examination under a microscope
takes only a few seconds or minutes, yet can identify unsuitable
crystals that might otherwise occupy hours on a camera or diffractometer.
A microscope with a polarising attachment, up to x40 magnification,
a good depth of field and a strong light source is required.
Crystal examination consists of three steps.
STEP ONE:
With the analyser component of the
polarising attachment out ( i.e. not in use)
look at the crystals in normal light to determine if they are
well-shaped. Reject crystals that are curved or otherwise deformed,
have significant passengers which cannot be removed, or that show
re-entrant angles. Be wary of rejecting crystals simply on the grounds
that they are small, unless similarly sized crystals of the same type
of compound have not previously been successful. For organic compounds
containing no element heavier than oxygen, crystals smaller than 0.1 x
0.1 x 0.1 mm3 seldom give good data with conventional
laboratory instruments, although this size may be ideal for crystals
of an osmium cluster compound.
STEP TWO:
With the analyser in, most crystals
in a typical sample will transmit polarised light. The exceptions
are tetragonal and hexagonal crystals viewed along their unique
c axis, and cubic crystals viewed in any orientation.
Tetragonal or hexagonal crystals transmit polarised light when viewed
along other directions but cubic crystals cannot be distinguished
from amorphous materials such as glass by this method. Fortunately,
these three crystal systems together account for less than 5% of molecular
crystals.
STEP THREE:
If a crystal transmits polarised light,
turn the microscope stage until the crystal turns dark (extinguishes),
then light again, a phenomenon that will occur every 90o.
This extinction is the best optical indication
of crystal quality, and it should be complete throughout the
crystal and be relatively sharp (ca. 1o).
Any crystal that does not extinguish completely is not single
and can be rejected immediately. Lack of sharpness may indicate
a large mosaic spread within the crystal. A crystal that never
extinguishes is almost certainly an aggregate of smaller crystals.
When examining a batch of crystals, establish both the general quality
of the sample and whether there are individual crystals suitable
for further study.
2.
X-ray photography.
Photography began to decline in popularity
as data collection using four-circle instruments advanced,
in part because it is often quicker to record a full dataset
is than to obtain a complete set of photographs. However, it is worth
remembering that photography gives a better view of the reciprocal
lattice than can be obtained from the list of reflections output
by a four-circle diffractometer, and can record any diffraction occurring
at other than the expected positions. An oscillation photograph
taken using a Polaroid cassette can be obtained within 5-10 minutes.
As well as giving information on crystal quality, photographs can
be used to establish unit cell dimensions and diffraction symmetry.
When screening crystals of dubious quality on a four-circle diffractometer,
there is probably no faster method than Polaroid photography.
The spread of area detector instruments will
finally consign X-ray photography to history. Area detector
images give much the same view of the reciprocal lattice as film,
but do so much more quickly, flexibly and precisely without the
need to process film.
3.
Diffractometry.
The ultimate test of a crystal is how
it behaves on the diffractometer. Reflections must possess
sufficient intensity, be well-shaped (not split or excessively
broadened) and index to give a sensible unit cell. Area detector
instruments combine some of the best features of photographs and
electronic counters and some can establish the quality of a crystal
in seconds. It is worth bearing in mind that area detectors can often
tolerate lower quality crystals than four-circle instruments, so
that crystals which would be inadequate for data collection on a
four-circle may be viable when using an area detector.
Standard procedures.
For crystals that are stable to ambient
conditions of air, moisture and light the requirements of
mounting are simple. The crystal is fixed securely with a reliable
adhesive (e.g. epoxy resin) onto a glass or quartz
fibre that is in turn glued into a "pip" which fits into the
well at the top of the goniometer head. The aim is to ensure that the
crystal does not move with respect to this head. This means rejecting
adhesives that do not set firmly ( e.g. vaseline or Evo-Stik)
or mounting media that are not rigid ( e.g. plasticine, Blu-tack
or picene wax). On some diffractometers crystals are spun at up to
4000o/minute, and an insecure mounting will lead to
serious problems of crystal movement.
A suitable fibre (e.g. of Pyrex glass) is just
thick enough to support the crystal at a distance of about 5 mm above
the pip. Fibres that are too thick add unnecessarily to errors
via absorption and background effects, while those that are
too thin can allow the crystal to vibrate, especially if it is being
cooled in a stream of cold gas. For normal-sized crystals, the fibre
should be thinner than the crystal. For small or thin crystals use
a "two-stage" fibre, which consists of a glass fibre onto which is
glued approximately 1 mm of glass wool to which the crystal is attached.
The fibre confers stability while the short length of glass wool
reduces the amount of glass in the X-ray beam.
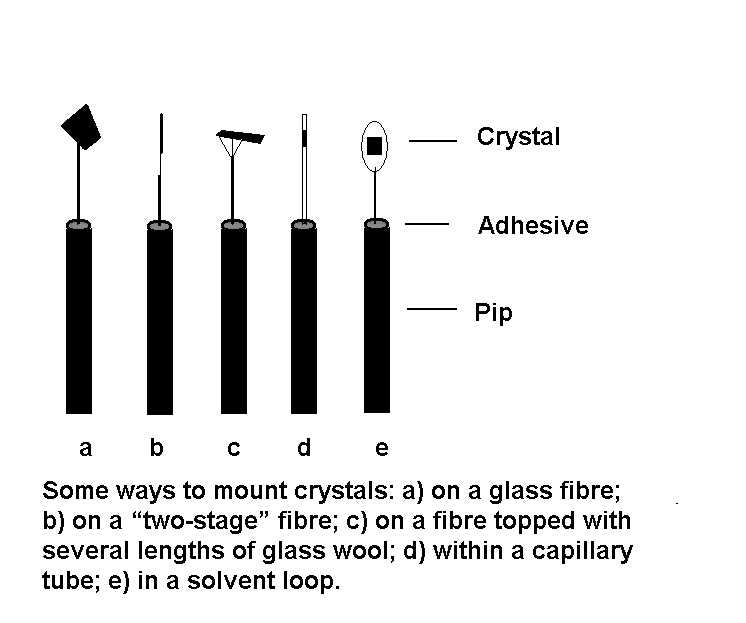
This paragraph outlines the basic procedure
for mounting a crystal. First mix the epoxy resin which
will typically become tacky within five minutes and thereafter
remain useable for a further five. Place the tip of the fibre
into the resin and use the microscope to check that it has actually
become coated. Ideally, the aim is to attach the tip of the fibre
to the side of the crystal, thereby minimising the amount of
glass in the X-ray beam. Establish the size of the crystals, cutting
them to size with a scalpel or razor blade if necessary.
When picking up a crystal there is a danger of
gluing it onto the slide, but this can be easily avoided. Move
the adhesive tipped fibre forward until it makes contact with the
side of the crystal, then continue moving the fibre forward and
upwards to lift the crystal clear of the slide. [With thin plates
this procedure may not be possible. If there is no alternative
to mounting a crystal with a fibre along an edge or across a face
of the crystal the fibre must be as thin as possible: a "two-stage"
fibre may be appropriate.] When picking up a crystal for use on a
four-circle instrument ensure that no crystal axis aligns perfectly
with the fibre (and therefore with the diffractometer φ axis) as this
can enhance systematic errors due to Renninger and other effects.
Also make sure that the crystal height can be adjusted to bring it
into the X-ray beam (it is frustrating to find later that this cannot
be done due to a fibre that is too long or too short – on many instruments
the X-ray beam passes 68 mm above the upper surface of the φ circle).
Instead of a simple fibre, some crystallographers prefer to mount
the crystal on the end of a capillary tube (less glass in the beam
for the same diameter); on a number of short lengths of glass wool
attached to a thicker fibre (ditto); or on quartz fibres (more rigid
for a given diameter).
Air-sensitive crystals
The traditional way to protect sensitive
crystals was to seal them (using a flame or epoxy resin) into
a capillary tube, usually made from Lindemann glass which is
composed of only light elements. Even so, this puts
a large volume of glass in the X-ray beam, so the tube and wall diameters
should be as thin as practicable. The most sensitive crystals
can be handled and encapsulated within a dry box. When planning
a low temperature data collection, ensure that the top end of the tube
is well rounded and that there are only a few millimetres of glass
above the crystal position, otherwise severe icing will result (alternatively,
see the second paragraph following). Crystals that desolvate may
need either solvent vapour or mother liquor sealed into the tube with
them. Unless crystals are mechanically robust, care must be taken
when loading them into capillary tubes. With crystals that are both
fragile and susceptible to solvent loss, a variant of a technique used
by protein crystallographers may be helpful. Break the sealed end off
a capillary tube and coat the first few millimetres of its inner surface
with freshly-mixed epoxy resin; place some crystals with their mother
liquor in a well and isolate a good crystal; bring the open end of the
tube through the surface of the solution; it may take some practice,
but capillary action should draw the crystal along with some mother
liquor into the tube; the crystal will stick to one side of the tube,
which can then be sealed at both ends.
Many crystals can be protected by coating
them with materials such as nail varnish, superglue or epoxy
resin. As long as the coating confers sufficient protection
and does not react with the crystal, this can be a simple and
effective solution to air-sensitivity that is applicable when cooling
of the crystal is impossible. This situation can arise because a phase
change is known or suspected to occur below ambient temperature, or
because cooling causes an unacceptable degree of mechanical strain within
the crystal.
A low temperature device permits the
use of an extremely flexible method for handling air-sensitive
crystals. This involves transferring, examining and mounting
the crystal under a suitably viscous oil. Upon cooling, the
oil forms an impenetrable film around the crystal and also acts
as an adhesive to attach the crystal firmly to the fibre. For crystals
that do not survive room temperature, the technique can be combined
with low-temperature handling, which normally involves passing
a stream of cold nitrogen gas across the microscope stage. Various oils
have been used but perfluoropolyethers have the advantages of inertness
and immiscibility with solvents. The excellent Riedel de Haen RS3000
is no longer produced, but one alternative is PFO-XR75, available
from Lancaster Synthesis, although it is not quite so inert and has
a lower viscosity. For many crystals silicone grease will be an adequate
substitute. However, if high viscosity is important try
http://www.abcr.de and do a chemical search for F06206R or
perfluoropolyether.
An alternative method, popular with
protein crystallographers and suitable for very thin crystals
that are too fragile to be picked up on a fibre, is the solvent
loop. A small loop of a fibre such as mohair or a single strand
from dental floss is used to lift the crystal in a film of solvent
or oil that is then flash cooled on the diffractometer to immobilise
the crystal. (For more details see E.F. Garman & T.R. Schneider,
Journal of Applied Crystallography
1997, 30 , 211-219.)
The final step is to attach the goniometer
head to the φ circle of the diffractometer and optically adjust
the crystal so that its centre does not move when it is rotated.
Do not assume that the microscope cross-hairs represent the true centre,
although if the instrument is reasonably well set up this should
be a useful starting point. Centring is an iterative procedure,
and the following outline should be possible on most instruments
with Eulerian cradle geometry:
Ø
First, with χ at 0o, make
sure the crystal is approximately central in X and
Y by checking at φ = 0, 90, 180 and 270o
then set the height Z approximately.
Ø
Second, view the crystal at φ 0 and
180o, then at 90 and 270o
. At any position the lateral offset must be the same as that
180o away.
Ø
Third, view the crystal at χ –90 and +90o.
Z is correct if the offset is the same at
each position.
Ø
Fourth, with χ at 0o re-check the
crystal at φ 0, 90, 180 and 270o.
Ø
Repeat the third and fourth steps until
convergence is achieved.
The above procedure will require adaptation
for different instrument geometries. For example, on a
Nonius CAD4 diffractometer X and Y are
checked at κ = –60o, and Z at κ = ±135o.
On instruments with fixed χ circles the height can
be checked by rotating ω through 180o.
Some References on Crystal Growth and Handling
Peter G. Jones, "Crystal
Growing", Chemistry in Britain 1981, 222–225.
[This article also covers
aspects of crystal evaluation and is highly recommended.]
H.E. Buckley, "Crystal
Growth", Wiley (London), 1951.
[Very detailed, good
for background and a source of alternative ideas for growing crystals.
Strangely, there is almost no mention of sublimation.]
T. Köttke and D. Stalke,
Journal of Applied Crystallography 1993, 26
, 615–619.
[The paper on the
use of oil films for handling sensitive crystals. Excellent on practical
aspects].
P.M. Dryburgh, B. Cockayne
and K.G. Barraclough (eds.), "Advanced Crystal Growth",
Prentice Hall International (UK) Ltd, 1987. (ISBN: 0-13-011249-6).
Look at the literature
on related compounds. [At the very least, the authors should have identified
the solvent they used and the temperature at which crystals
were grown.]
Crystal growing hints and tips on the
Web
http://www.xray.ncsu.edu/GrowXtal.html
http://www.cryst.chem.uu.nl/lutz/growing/gel.html
http://www.cryst.chem.uu.nl/lutz/growing/reading.html
http://www.cryst.chem.uu.nl/growing.html
Top