{kind=link}
{kind=link}
3.3- Solving the structure
At this point of the scenario, the next step is the same one as if we had made the study of a very bad single crystal with a four-circle diffractometer. Indeed, we have quite bad data at our disposal. This is easily understandable. The more a reflection has close neighbouring reflections, the more the "observed" structure factor "|Fobs|" is dubious. In practice, the exact overlapping of two reflections leads to the following proposal : the two reflections have the same "|Fobs|" . The Le Bail method applies the Rietveld decomposition formula starting from a set of all identical |Fhkl|s replacing the |Fcalc|s in the normal Rietveld method. As the repartition to the "|Fobs|" in a group of reflections is made according to the |Fcalc|, then two reflections with the same position will receive the same partition and so will remain equal up to the end of the iterative process : this is the so called equipartition. Of course, this is false, however this is the most acceptable proposition we can do at this stage. In fact some programs (as GSAS) offer to start from |Fhkl|s calculated from a dummy atom, this is to be avoided because the strictly overlapping reflections will no be equipartitionned, so that the final dataset will keep some memory of the dummy atom ; moreover, the null reflections will remain at zero after each iteration, even if there is observed intensity (it is thus adviced to choose the dummy atom at a general position, not at a specail one which could correspond to special extinctions). Maybe it would be possible to redistribute the equipartitionned intensities at a next stage by some special methods proposing to use the information contained in the isolated reflections in order to differenciate the superposed reflections. A less sophisticated means which has proved its utility consists in the elimination of the more dubious reflections from the dataset by using a small software (OVERLAP). If the direct methods are needed, then one should not suppress more than 50% of the reflections. If only few atoms have to be found by the Patterson method then one can suppress more than 50% keeping some chance to obtain the required starting structural model. It would be preferable to associate the criterium for eliminating reflections to the FWHMs rather than to use a fixed proximity value. However the OVERLAP software simply eliminate a reflection if it has a neighbouring one at less than X° 2-theta. If the application of the direct and/or Patterson methods is unsuccessful with the complete dataset, then the game consists in applying them to reduced datasets with X = 0.01 and then 0.02, 0.03, 0.04° 2-theta, etc. Expecting to attain the limits given at chapter 3.1.1. one should not use X larger than 0.02° 2-theta.
The choice of the method (direct, Patterson) is suggested by the presence or not of "heavy" atoms. There is no difference here with the strategies recommended for a single crystal study. In this scenario were tested the well known programs SHELXS86/SHELX76 (SHELXS86 has been updated and is now inside the SHELX97 package together with an updated version of the SHELXL93 refinement program). You will not find here the manuals of these programs, you will have to read and assimilate them completely, playing with the test files.
The optimal conditions for determining a structure from the direct methods (when the initial model for starting a refinement will be larger than 2 or 3 independent atoms) correspond to a 1 Å resolution (2-theta max ~ 100° for a Cu Kalpha wavelength). No more than 50% of the reflections corresponding to this resolution should be discarded by the above OVERLAP program. Frequently, the structure is determined by using the whole dataset. This may be considered amazing when the number of false structure factor is as high as 50% or evenmore. One can think that the false structure factors are randomly false so that the process which consists in searching for order is not affected. The consequence of random errors will be to increase a background above which the peaks associated with the order may still be located. It will be however more and more difficult to find these order-peaks when the true information is more and more diluted.
Finally, some words about the space group. Sometimes it is really impossible to have a non ambiguous proposition. In the worst cases, several groups may present the same extinctions. Among the possible space groups you should always test the group with the highest symmetry (unless you have external informations, for instance a positive test of acentricity).
We will continue with the scenario samples :
Na2C2O4
No heavy atom in this compound, so the direct methods
will be applied by the use of SHEXS86 (don't forget to read the manual
carefully ). In a first approach, the complete dataset (325 "|Fobs|")
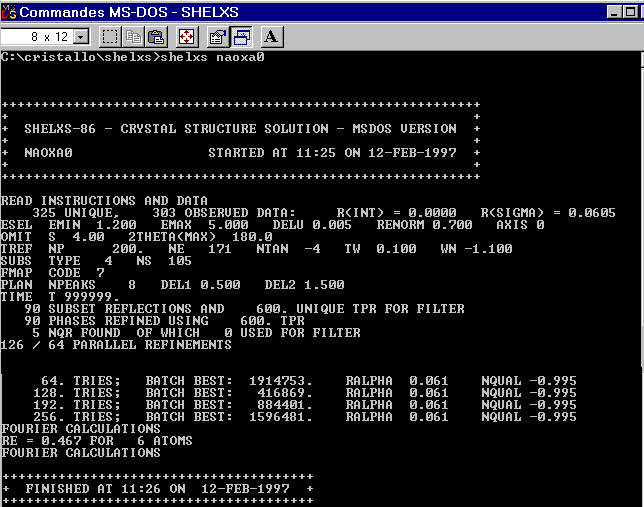
will be retained. SHELXS86 is started easily in a DOS box (figure
24). The datafile is shown in naoxa9.html
together with the result. You should take your time and first of all you
should compare this result with those obtained from reduced datasets as
prepared by the OVERLAP program. Eliminating
the reflections having a neighbouring one at 0.02° and 0.04° 2-theta,
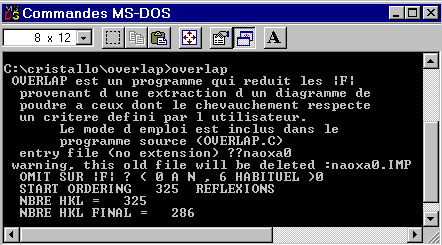
the remaining reflections are respectively 286 and 242 hkl. OVERLAP is
easy to run, see the figure 25. The summary
of the three above attempts corresponding to the five first atoms in the
list proposed by SHELXS86 is in naoxa10.html.
The next step will be the eventual validation of one of these propositions
or of some atoms in the list.
[Pd(NH3)4]Cr2O7
For this compound, the Patterson method is the best choice,
in principle. Nevertheless the direct methods remain efficient when a small
number of atoms have to be found. With SHELXS86, a Patterson followed by
an automatic search is much longer in computer time than a direct methods
application. That is why my first try is by the direct methods in any case.
However for the demonstration purposes, we will follow first the more logical
way : the Patterson method. This time, with more than 1000 reflections,
and according to the fact that the FWHMs are relatively large, few "|Fobs|"
are really well estimated. The Patterson method can be applied from a very
limited dataset when the number of "heavy" atoms to be located
is small. An application of the OVERLAP program with X = 0.08° 2-theta
produces a dataset reduced to 137 reflections. The PATT option of SHELXS86
proposes two atoms : Pd at 0,0,0 coordinates and
Cr at 0,0,0.5. The chromium atom being expected in a tetrahedral environment
should not occupy an inversion center. We may believe to know the formula
and attribute 20 A3 per NH3 group and per oxygen
atom : the total is 11x20 = 220 A3 per formula unit to be compared
to a cell volume slightly less than 1000 A3 so that the number
of formula units per cell could be Z = 4. If the formula is exact, the
two sites proposed by SHELXS86 should be two palladium atoms sites. A first
refinement (with SHELX76), leads to a result quite
unsatisfying with R = 0.62. However the Fourier difference synthesis proposes
2 peaks clearly distinct in weight which could be associated with 2 independent
chromium atoms. This hypothesis is tested with SHELX76. The reliability
factor goes down to 35% and it seems that the Fourier
difference synthesis continue to propose acceptable sites (at least
for the three peaks in head of the list) which could correspond to oxygen
atoms (being at 1.61 or 1.57 A near of the supposed Cr atoms) or N atoms
(being at 2.19 A from a supposed Pd atom). The job consists in introducing
the new atoms and to refine until nothing more is recognized on the Fourier
difference synthesis. And what would have given the direct methods ? Application
to the 1054 hkl, of which probably no more than 250 are more or less well
estimated, proposes randomly the 4 heavy atom sites (2 Pd + 2 Cr) at the
head of the list and evenmore !
t-AlF3
The brute force consisting in the SHELXS-97 application
to the whole dataset gave a list of 15 atom sites (talf33.html).
Examining this list by a structure drawing program (STRUVIR) revealed that
the 11 first atom sites described completely a new MX3 corner
sharing 3D network. Where is the merit of the researcher ? Thanks to G.
Sheldrick !
beta-BaAlF5
From neutron data, we have no other choice than to apply
the direct methods. The more complex case previously
solved exclusively from neutron data allowed to locate 6 independent atoms.
Here we have 14 sites to locate from low resolution data ! Proceeding meticulously,
that's the way. Several datasets were prepared corresponding to different
X values in the OVERLAP program. All these datasets were used as hkl files
for the direct method application in SHELXS-97. The 14 first peaks of the
direct method propositions were introduced into the SHELX76 program, irrespectively
of their possible meaning (attributing to all atoms the Ba Fermi length)
and a refinement was made on a reduced dataset (338 hkl corresponding to
X = 0.06). Wen all the datasets were treated, the whole result was observed
:
X in OVERLAP Dataset R(%)SHELX76 0 1387 36 0.01 1099 51 0.02 866 39 0.03 691 39 0.04 522 46 0.05 427 46 0.06 338 42
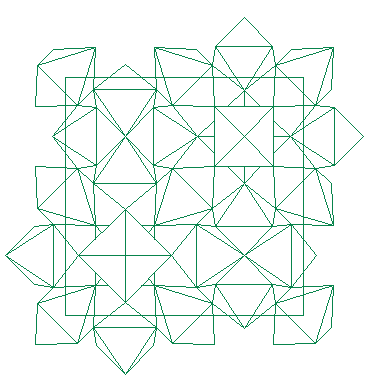
Curiously, the best proposition (R = 36 %) is obtained from the whole dataset. In fact the data were limited to the 1266 first reflections in order to limit the maximum h,k,l values (6, 22, 8) to just a little more than the a, b, c parameters (~ 5, 19, 7), as recommended for applying direct methods (going to dhkl ~ 1 A). See the SHELXS-97 proposition in bafef53.html and the SHELX76 refinement result in bafef54.html). Looking more accurately to this proposition, it becomes clear that the Q12 site could be an Al atom in octahedral coordination (with Q1, Q3, Q6, Q8, Q9 and Q11 sites as F atoms). The Q13 site presents short distances with Q3, Q4 and Q7 sites, however it is at 2.40 A from Q12 so that it seems to be an artifact. Nevertheless, a structure drawing by STRUVIR is very convincing that at least a large part of the beta-BaAlF5 structure has been obtained. Other interatomic distances could be acceptable as F-F or Ba-F distances. The AlF6 octahedra (see below) could not be artifact, in spite of the fact that a correct proposition from the whole dataset was certainly unexpected due to the low resolution data.
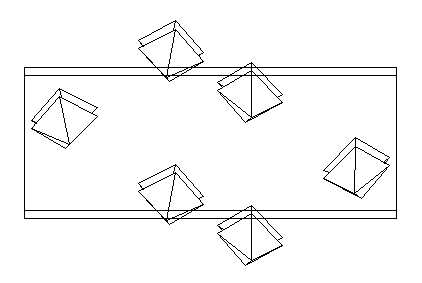
The next step will be obviously to apply the Rietveld method with 13 of the proposed atom sites (excluding Q13). This seems to be a new example that equipartitionning the overlapping reflections produces a random error which does not perturbate the direct methods searching only for order. Of course, solving the structure from the X-ray data was much easier. The two barium atoms only had to be located. They were sufficient to start refinement and find the remaining atoms by Fourier difference synthesis.
Cimetidine C10H16N6S
In the previous structure redetermination from synchrotron powder
data (J. Appl. Cryst. 24, 1991, 222-226), the authors input the data extracted
by the Pawley method into a number of direct-methods programs, all
of which failed to solve the whole structure but correctly located 3 to
4 atoms including the sulfur. The structure was finally solved by taking
the top 17 peaks from the output of the direct-methods program SIR. Here,
the 924 reflections extracted by the Le Bail method are put into
SHELXS-97 applying the direct methods by the "brute force". The
resulting top 17 peaks were introduced into SHELXL
for a refinement against a reduced dataset (OVERLAP application with X
= 0.03, 378 remaining reflections) more or less irrespective of their physical
sense with an exception for the first peak designed as the S atom. A very
encouraging R1 ~ 18% was obtained allowing
to go further. Examining interatomic distances and the molecule permitted
to distinguish between N and C atoms and to attain the next stage consisting
in a Rietveld refinement.